Protocol can take from 30 minutes - 40 minutes by QIAGEN
Cells: Harvest a maximum of 5 x 105 cells, as a cell pellet, or by direct lysis in the vessel. Add 350 µl Buffer RLT and homogenize.
Tissues: Disrupt and homogenize ≤5 mg tissue in 350 µl Buffer RLT using the TissueRuptor® or TissueLyser instruments. Centrifuge the lysate for 3 min at maximum speed. Carefully remove the supernatant by pipetting and use it for step 2.
Microdissected cryosections: Collect the sample directly into an appropriate volume of Buffer RLT. (The volume depends on the collection vessel used for microdissection, but should not exceed 65 µl [Leica® instruments] or 300 µl [other instruments].) Adjust the volume to 350 µl with Buffer RLT. Vortex for 30 s.
Add 1 volume of 70% ethanol to the lysate, and mix well by pipetting. Do not centrifuge. Proceed immediately to step 3.
Transfer the sample, with any precipitate, to an RNeasy MinElute spin column in a 2 ml collection tube (supplied). Close the lid and centrifuge for 15 s at ≥8000 x g. Discard the flow-through.
Add 350 µl Buffer RW1 to the RNeasy MinElute spin column. Close the lid. Centrifuge for 15 s at ≥8000 x g. Discard the flow-through.
Add 10 µl DNase I stock solution to 70 µl Buffer RDD. Mix by inverting the tube. Add the DNase I incubation mix (80 µl) directly to the RNeasy MinElute spin column membrane. Place on the benchtop (20–30°C) for 15 min. Add 350 µl Buffer RW1 to the RNeasy MinElute spin column. Close the lid, and centrifuge for 15 s at ≥8000 x g. Discard the collection tube.
Place the RNeasy MinElute spin column in a new 2 ml collection tube (supplied). Add 500 µl Buffer RPE to the spin column. Close the lid, and centrifuge for 15 s at ≥8000 x g. Discard the flow-through.
Add 500 µl of 80% ethanol to the RNeasy MinElute spin column. Close the lid, and centrifuge for 2 min at ≥8000 x g. Discard the collection tube.
Place the RNeasy MinElute spin column in a new 2 ml collection tube (supplied). Open the lid of the spin column, and centrifuge at full speed for 5 min to dry the membrane. Discard the flow-through and collection tube.
Place the RNeasy MinElute spin column in a new 1.5 ml collection tube (supplied). Add 14 µl RNase-free water directly to the center of the spin column membrane. Close the lid gently, and centrifuge for 1 min at full speed to elute the RNA.
This protocol below is taken from a protocol textbook
Isolation of High-molecular-weight DNA from Mammalian Cells using Proteinase K and Phenol
Extraction of DNA from lysate
Transfer lysate to one or more centrifuge tubes. The tubes should not be more than one-third full.
Add proteinase K to a final concentration of 100 µg/mL to each centrifuge tubes. Using a glass rod, mix the enzyme solution gently.
Incubate lysate in a water bath for 3 hours at 50 ˚C. Swirl the solution from time to time.
Cool the solution to room temperature and add an equal volume of phenol equilibrated with 0.1 M Tris-Cl (pH 8.0). Gently mix the two phases by slowly turning the tube end-over-end for 10 minutes on a tube mixer or roller apparatus. If the two phases have not formed an emulsion at this stage, place the tube on a roller apparatus for 1 hour.
Separate the two phases by centrifugation at 5000g (I don’t know what centrifuge we’ll use so I don’t know what RPM) for 15 minutes at room temperature.
Use a wide-bore pipette to transfer the viscous aqueous phase to a fresh centrifuge tube.
When transferring the aqueous phase (usually the upper phase), it is important to draw the DNA into the pipette very slowly.
Repeat the extraction with phenol twice more and add them to previous aqueous phases.
Isolate DNA
DNA Isolation in the size range of 150-200 kilo-bases
Transfer aqueous phase to a dialysis bag, and allow the bag to increase 1.5-2 fold during dialysis.
Dialyze the solution at 4 ˚C against 4 liters of dialysis buffer. Change the buffer three times at intervals of ≥ 6 hours. Typically dialysis takes ≥24 hours to complete.
DNA Isolation in the size range of 100-150 kilo-bases
After the extraction, transfer the aqueous phase to a fresh centrifuge tube and add ⅕ of the volume of 10 M ammonium acetate. Add 2 volumes of ethanol at room temperature and swirl the tube until the solution is thoroughly mixed.
DNA Precipitate should form. Remove the precipitate in one piece using a shepherd’s crook.
If the precipitate is fragmented then centrifugate at 5000g for 5 minutes at room temperature
Wash the DNA precipitate twice with 70% ethanol.
Remove the 70% ethanol as much as possible and allow it to dry but not completely dry.
Add 1 mL of TE buffer (pH 8.0) for each 0.1 mL of cells in step 1. Place the tube on a rocking platform and gently rock the solution for 12-24 hours at 4 ˚C until the DNA has completely dissolved. Store the DNA solution at 4 ˚C.
Meanwhile, the protocols here are more convenient to use as this will allow us to purchase an extraction kit.
Protocol can take from 20 minutes - 1 hourby QIAGEN
Place sample into a collection microtube.
Prepare a working solution containing 20 µL of proteinase K stock solution and 180 µL buffer ATL per sample, and mix by vortexing. Immediately pipet all into each collection microtube. Tightly seal the microtubes using the caps provided.
Place the clear cover over each rack, and mix by inverting.
Incubate at 56 ˚C overnight or until the samples are completely lysed. Place a weight on top of the caps during the incubation. Mix occasionally during incubation.
Cover the racks and vigorously shake for 15 seconds. Ensure that samples are completely lysed to avoid clogging wells of the DNeasy 96 plate.
Remove the caps and add 410 µL buffer AL-ethanol mixture to each sample, and tightly reseal using new caps.
Place a clear cover over each rack and shake the racks vigorously for 15 seconds.
Place 2 DNeasy 96 plates on top of S-Blocks. Mark the DNeasy 96 plates for later sample identification.
Carefully remove microtube caps and transfer the lysate of each sample to each well of the DNeasy 96 plates.
Seal each plate with an AirPore Tape Sheet. Centrifuge for 10 min at 3800g.
Remove the tape. Add µL buffer AW1 to each sample.
Repeat step 10 with a new tape sheet and centrifuge for 5 minutes at 3800g.
Add µL buffer AW2 to each sample
Centrifuge for 15 minutes at 3800g.
Place each DNeasy 96 plate on a new rack of Elution microtubes RS.
Add 200 µL buffer AE to each sample, and seal with new AIrpore tape sheets. Incubate for 1 min at room temperature. Centrifuge for 2 min at 3800g.
Seal the elution microtubes RS with new caps to store the eluted DNA.
DNA Extraction in 15 minutesby Thermofisher Scientific
Nucleic Acid Extraction by
Mechanical Means
Cell Disruption
Cell Disruption, the first step in extraction normally requires a centrifuge. But, it seems there are other mechanical/chemical methods out there that can disrupt the cell.
"Cell disruption or disintegration can be achieved by physical and/or chemical methods, whose main aim is to disrupt the cell wall and/or cellular membranes. Disruption methods are mainly based on properties of the sample and for this purpose a wide range of tools and approaches are used either alone or combined to achieve tissue/cell disruption [7]. Lytic enzymes, chaotropic agents, and different types of detergents are the main components of chemical lysis, while mechanical method disrupts the cells by grinding, shearing, bead beating, and shocking [8]. It is interesting to note that if one technique does not yield good results, another might prove successful. Osmotic shock methods have yielded, in certain cases, better results than common NA purifications protocols such as phenol-chloroform extraction and bead beating [9]."
Reaches temperatures to 75 and 95 celcius. In addition to high temperatures, a cocktail of enzymes and other chemicals are used to lyse the cells and extract the nucleic acid from the cells.
Almeida da Silva et al. 2012 .This method employs a centrifuge-less DNA extraction method by boiling and freeze-thawing.
However, this method requires to expose the sample in extreme temperatures: 90 °C and -18 °C. Most probably impractical.
Bead Beating
Bead beating is a mechanical method of disrupting biological samples. Essentially, bead beating is accomplished by rapidly agitating a given sample in grinding media i.e. bead water, as a result, breaking open the cell containing the target DNA. - Islam et al. 2017
This OmniLyse product can lyse cells, including fungal cells within 1 minute using a bead beating method.
This is the earlier methods employed for cell disruption. Utilizing high pressure, the french pressure cell press is essentially a hydraulic press that forces cells through a narrow crevice, ultimately lysing the cells due to the shear forces created when pressed. - Islam et al. 2017
Using sound energy, ultrasonication is an alternative method to agitate a sample of cells, ultimately leading to cell lysis. Ultrasonication typically requires frequencies of at least 20 kHz to function. - Islam et al. 2017
Updated by @Yaman Garg
Portable Ultrasonication for Eukaryotic human leukemia HL-60 cells (80% lysis in 3s) and Bacillus subtilis bacterial spores (50% lysis in 30s) was described by Marentis et al., 2005 where they made a microelectromechanical-based piezoelectric microfluidic minisonicator operated in the 380 MHz range with the following device design where the solution is passed through the channel milled in the glass substrate for lysis and the electrodes and piezo-electronic transducers fabricated on the quartz substrate create the resonance:
Powering: The transducers were driven by a continuous sinusoidal power source (AC) in the 360-MHz range and a 20dB amplifier in the experiments to create resonance/sonication, the maximal peak to peak voltages observed were 4.5 V (human cells) and 10 V (bacterial spores), with powers 0.2 W and 1 W respectively.
A Problem: The authors also mentioned a heating problem "During a 33-dBm sonication test, the temperature in the channel increased from 20°C to 70°C with 5 μL/min flow and from 20°C to 65°C with 50 μL/min flow for a 3-s residence time." which they solved by using an ice pack.
Salehi-Reyhani et al., 2015 described a surface acoustic wave (SAW) based device for for 30 second lysis of BE human colon carcinoma cell to capture p53 protein which worked by creating vortexes in stationary droplet (20 μL) of cell culture. However, at 2W most nuclei remain intact or the DNA remains compacted and the efficiency of SAW-induced mechanical lysis was determined to be around 10% of that for conventional detergent-based lysis in yielding detectable protein.
SAWs were generated by piezoelectric 128° Y-cut X-propagating 3-in LiNbO3 wafers. The devices consisted of 20 pairs of electrodes to form an interdigitated transducer (IDT). Frequencies with which vortices were achieved in the droplet varied between 9.7 and 10.3 MHz.
The authors described a phononic superstrate which could be detached from the expensive IDT + LiNbO3 system and worked as disposable sample holder. The superstrate was placed on top of the piezoelectric wafer and coupled with 2 μL of water-based gel (KY jelly; Johnson and Johnson) spread manually in between, yielding a film approximately 50 μm thick. It comprises a square array of circular holes in a 470 μm thick silicon wafer that scattered the SAW to obtain an asymmetry in the acoustic field. An area of 5 mm in diameter on the surface of the superstrate was made hydrophilic at the location where the sample droplet would be located. The rest of the surface was made hydrophobic.
The device design is as follows:
The device was run by an MXG analog signal generator N5181A (Agilent Technologies) in conjunction with a Mini Circuits ZHL-5W-1, 5–500 MHz amplifier.
The lysis was also performed on droplets of cells containing an equal concentration of beads which really improved the process, at 2W (-4 dB) cell nuclei were predominantly disrupted with a few remaining intact, still not much efficiency gain.
Reboud et al., 2012 had used the device described above for lysis of blood cells with malarial parasites with 98% efficiency at a power of 0.8 W in 4 secs. Fun fact: they also used SAW for PCR (for heating) (and a thermal sink for passive cooling).
Taller et al., 2015 also describe a SAW lysis device to extract miRNA from exosomes, which lysed 100uL in 30 mins with a rate of 40-50% exosomes lysed with a power of 1 W. Process diagram:
The SAW device was fabricated using standard UV photolithographic methods. Twenty pairs of titanium/aluminum interdigitated electrodes were patterned on a 127.68° yx-cut piezoelectric lithium niobate (LiNbO3) substrate (16 mm × 40 mm and 0.5 mm in thickness) to make a SAW transducer (SPUDT), which generates plane SAWs propagating in one direction only. The operating frequency was 28.3 MHz. The SAW was activated by a function generator (Agilent 33250A) in series with an amplifier (E&I 325LA RF Power Amplifier). A channel for fluid flow was fabricated using three layers of polycarbonate thermosoftening plastic. Device design:
This kit claims to be able to extract and purify DNA within 1 minute. There really isn't much info on the exact details of the kit. But according to them, they use a lysis buffer to lyse the cells and the use of a silica membrane to selectively bind DNA and filter any unwanted material.
Based on the principle that DNA is negatively charged, a positively charged silica membrane is used to selectively bind DNA/RNA. According to this paper, the rest of the cellular contaminants can be washed out using distilled water or a buffer like Tris-EDTA. Major limitation: It cannot be reused!
Used to maintain a suitable pH for target molecules
Salts
Used to maintain ionic strength. This is normally used for protein purification to maintain structural integrity.
Detergents
An amphipatic molecule that disrupts the cell membrane of the target cell. This is the main ingredient and the strength of your detergent determines the strength of your lysis buffer. Common detergents: Triton 100 - X or SDS (Sodium Dodecyl Sulfate); the latter is a much stronger detergent.
Chelating Agent
These are chemicals that react with metal ions like magnesium and calcium. This is mainly used to prevent any DNAse or Protease activity from happening. Common Chelating Agent: EDTA (Ethylenediaminetetraacetic Acid)
Note for table above: This is assuming 1 L of lysis buffer is prepared.^
Grand Total: $31.48 for 1 L
It seems that the proportions of the reagents used in a lysis buffer is largely dependent on the situation/cell type. The recipe above is taken from a paper that uses a lysis buffer on fungi, but not specifically Bd/Bsal.
Reagent Quantities:
According to Coloe et al. 2000 , 500 μl of lysis buffer is enough for a small lump of fungi taken from a toothpick. I doubt we would be collecting the same amount of fungi from a skin swab, so, at most, we would need 500 μl for cell lysis. Therefore, assuming we are using the recipe above, one test would cost $0.01574. very cheap wow. profit.
Lysing Cost Per Diagnostic Test: $0.01574
Storage Conditions: Can be stored in room temperature for over a year.
This is a common protease used to lyse the cells. In addition to proteinase K, SDS is also added to dissolve cell membranes and denature and unfold proteins. - Dairawan et al. 2020
"The mechanical methods appeared to be the most effective for the disintegration of yeast, R. gracilis, and filamentous fungi, A. fumigatus and P. citrinum. Ultrasonication and bead milling led to enzymatically active cell-free extracts, which can be a starting point for the isolation and purification of intracellular components of interest."
"Finally, the chemical treatment [detergent-based lysis] led to unsatisfactory results in every experimental attempt.In the case of charged detergents, there is also the possibility of a destructive impact which comes from the ionic interaction between enzyme and detergent residues."
Used to maintain a suitable pH for target molecules
Salts
Used to maintain ionic strength. This is normally used for protein purification to maintain structural integrity.
Detergents
An amphipatic molecule that disrupts the cell membrane of the target cell. This is the main ingredient and the strength of your detergent determines the strength of your lysis buffer. Common detergents: Triton 100 - X or SDS (Sodium Dodecyl Sulfate); the latter is a much stronger detergent.
Chelating Agent
These are chemicals that react with metal ions like magnesium and calcium. This is mainly used to prevent any DNAse or Protease activity from happening. Common Chelating Agent: EDTA (Ethylenediaminetetraacetic Acid)
Note for table above: This is assuming 1 L of lysis buffer is prepared.^
Grand Total: $31.48 for 1 L
It seems that the proportions of the reagents used in a lysis buffer is largely dependent on the situation/cell type. The recipe above is taken from a paper that uses a lysis buffer on fungi, but not specifically Bd/Bsal.
Reagent Quantities:
According to Coloe et al. 2000 , 500 μl of lysis buffer is enough for a small lump of fungi taken from a toothpick. I doubt we would be collecting the same amount of fungi from a skin swab, so, at most, we would need 500 μl for cell lysis. Therefore, assuming we are using the recipe above, one test would cost $0.01574. very cheap wow.
Reaction Time: 10 minutes
Lysing Cost Per Diagnostic Test: $0.01574
Storage Conditions: Can be stored in room temperature for over a year.
Cost: $75.30 for 500 mL ($0.1506/1mL) | $0.0753 per test
Buffer P2 is a lysis buffer used when purifying plasmid DNA. (Taken directly from site)
UPDATE: Aug 9
Enzymatic Lysis
The main purpose of using a lytic enzyme for cell lysis is to target the cell wall. All cells are built different, including their cell wall (if they have one). This makes lytic enzymes selective.
A common enzyme that is used in enzymatic cell lysis, especially amongst bacteria. Lysozyme mainly reacts with the peptidoglycan layer of the cell wall by breaking the glycosidic bonds of the polymers, ultimately brekaing down the cell membrane of the cell. It is important to note, however, that not all cells have the peptidoglycan layer, which according to my findings, include chytridiomycota (Bd/Bsal). That said, this will most likely not work on Bd/Bsal as the cell wall is mainly composed of chitin.
Optimum conditions of this enzyme is generally used in pH 6-7 and at 35 °C. This has been found to work on yeast.
Chitinase has been found to be involved in the decomposition of the fungal cell wall. Chitinases hydrolyze the linear polymer chitin, a polysaccharide of β-(1,4) linked N-acetylglucosamine (GlcNAc; 2-acetamino-2-deoxy-β-d-glucose) units. In other words, Chitinases break down chitin, the main material that makes up the cell wall of chytridiomycota.
It indicated an optimum activity in pH 5 at 45°C. Enzyme was stable in 55°C for 20 min and at a pH range of 3-9 for 90 min at 25°C.
The enzyme can be bought commercially, as indicated in the link above. For 50 units, it will cost $860. Their unit definition is: One unit will liberate 1.0 mg of N-acetyl-D-glucosamine from chitin per hour at pH 6.0 at 25 °C in a 2 hour assay.
A paper published in 1995 by Bir et al. 2007 that uses chitinase to extract DNA from Aspergillus Fumigatus (A fungi). Suspended in pH 6.0 in phosphate buffer, fungal cells were incubated in 0.5 U of chitinase per gram wet weight of cells at 25 °C for 90 minutes. Afterwards, the sample was incubated in detergent-based lysis buffer (They used SDS) for 90 minutes.
This whole process took 3 hours, making it unideal. Also, since they use both a detergent-based lysis buffer and a lytic enzyme, using chitinase alone may not be able to lyse a chitinous fungal cell alone. This may have to be tested in the lab. Most of the time, lytic enzymes are used in addition to detergent-based lysis buffers, most likely to improve lysis efficiency.
A useful figure that illustrates the efficiency of chitinase in different concentrations and incubation time
This is a lytic enzyme sold by Zymo Research that exhibits the following activities:
β-1,3 glucanase
β-1,3-glucan laminaripentao-hydrolase
Ultimately hydrolyzing glucose polymers at the β-1,3-glucan linkages. This is intended to be a lytic enzyme that targets fungal cell walls, including a lot of genera as indicated in the link. Although, it does not mention anything of being able to work on Chytridiomycota, Bd, nor Bsal.
This is an enzyme normally used for digesting proteins in nucleic acid purification. So, this enzyme doesn't actually accomplish any enzymatic lysis activity, rather inactivates any DNAse or RNAse activity very well to prevent targetted nucleic acids from breaking down.
Bead Beating
@Maya Fayed
"Molecular detection of microorganisms requires microbial cell disruption to release nucleic acids. Many common pathogens can be lysed through chemical agents, such as detergents and chaotropic salts, or by enzymatic treatment. However, lysis is a significant challenge for thick-walled microorganisms. High-energy mechanical disruption methods, such as bead beating, are commonly used for lysis of thickwalled organisms, since chemical, heat, freeze-thaw, or enzymatic lysis methods alone are less effective. This mechanism of lysis by bead-beating has been attributed to high shear rates between beads and strong periodic vortical flow fields"
Devices
The devices currently available for bead beating include:
The Mini-BeadBeater and the FastPrep Lysis Systems are among the smallest bead beating devices on the market large amounts of power (typically mains electricity), are less suited to poratble point-of-care testing.
OmniLyse and AudioLyse, however, offer low-power, low-cost alternatives to these benchtop bead beating devices. These devices are also disposable, more suited for commercial production and significantly less expensive than the Mini-BeadBeater ($2200) the FastPrep ($7000). A pack of 48 OmniLyse kits along with battery packs costs $540 and the components of one AudioLyse units would cost less than $40.
Mechanism and Components
OmniLyse:
The device consists of an injection-molded chamber with inlet and outlet ports and a lead-free motor that can operate at full speed for up to 20 minutes. It uses four AAA 1.5-V batteries, weighs approximately 3 g, and is 1.1 cm3 without a battery pack. The system is a single-use disposable unit and implements inexpensive fabrication methods through injection molding for the main body and fittings as well as mass-produced motors that can be obtained at a low cost.
The use of OmniLyse for the lysis of Bacillus subtilis spores and Mycobacterium bovis
BCG cells was described by Vandeventer et al., 2011 where they proposed the device as a disposable, battery-operated/ low-power, light weight, miniaturized, bead blender using the Biospec Mini-BeadBeater as a benchmark standard.
The authors operated the device with voltage of 6 V and an electrical current between 100 and 200 mA. For lysis using the OmniLyse device, 500 microliter sample aliquots were aspirated from the sample tube through the stiff tubing into the device and into a syringe attached at the port with the luer lock and then dispensed back through the device into the sample tube for a total of three passes, with a total processing time per sample of 1.5 min. The integrated micro motor is able to drive the impellor at speeds greater than 30,000 rpm with the chamber filled with liquid and beads where the kinetic energy imparted onto the beads in solution generates high shear forces between beads, causing disruption of cells caught within the shear flow. The liquid is then ejected from the device after 30 seconds and the beads are retained in the bead mill separating them from the lysate by the use of wire mesh filters. Crude lysates are kept on ice until samples are tested using PCR.
OmniLyse Isometric Schematic - The impeller can be clearly seen here, attached to the motor OmniLyse Patent
Note: a potential problem here is that the samples would need human intervention in between passes which would reduce time-efficiency. Additionally, the need for a battery back means that the device is harder to integrate into a lab-on-a-chip microfluidic device.
The OmniLyse device was found to mechanically lyse B. subtilis spores and M. bovis BCG cells with efficacy similar to the industry-standard benchtop BioSpec Mini-BeadBeater, based on the comparable CT values obtained from PCRs with primers specific for these two microorganisms.
AudioLyse:
Buser et al., 2015 propose an audio-powered mechanical lysis that utilizes low-cost, low-power equipment as an alternative to OmniLyse. The device demonstrates mechanical cell lysis through bead beating that is powered by an audio device (mp3 player in this case but any device with a headphone jack can be used) coupled with a simple and inexpensive electromagnetic coil. The magnetic field from the coil rotates the magnet inside a tube with the sample and glass beads enabling cell lysis for sample preparation.
Pros:
Freedom from additional power supplies (ex: battery packs in OmniLyse)
Potential for using the same equipment (cell phones) for both sample preparation and interpretation of results
Low cost (less than $38 was spent for the mp3 player, magnet wire, cable, and bobbin materials per device, and less than $1 per assay was spent for the screw-top tube, magnet, and beads)
Quieter technique more suitable to the point-of-care
Portable design
Cons:
Decreased Lysis efficiency
Longer process than OmniLyse (majority of amplifiable DNA is released in about 10 minutes)
Whether the efficiency of the device is suitable for us would have to be determined by infection levels, sampling technique, and the associated assay sensitivity
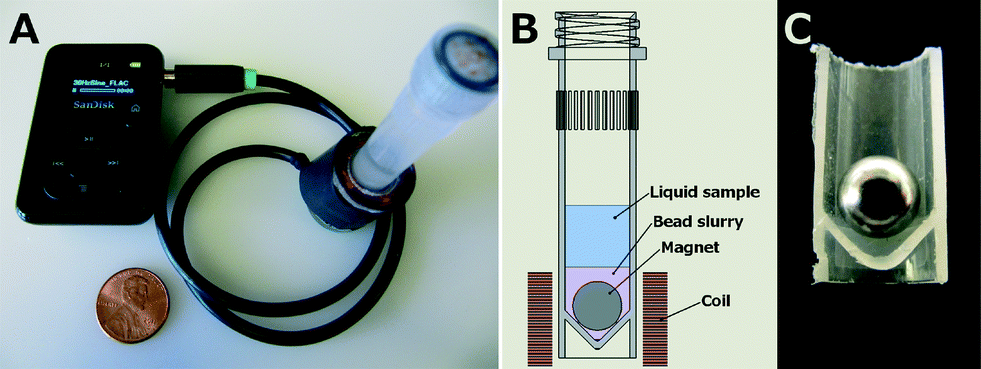
The device design is as follows:
"Overview of portable audio-powered electromagnetic cell lysis device. A: Sansa Clip mp3 player provides a 30 Hz sine wave signal to the coil using the headphone jack, US penny for scale. B: Schematic of tube showing user-added sample along with pre-loaded magnet and beads. C: Sectioned tube with magnet. The spherical magnet rides on the lower angled tube walls when the tube is vertically oriented. When provided an alternating magnetic field by the coil, the magnet rotates against the beads and the tube walls."
OmniLyse vs. AudioLyse
Heiniger et al., 2016 directly compare the bead beating for OmniLyse and AudioLyse by testing their ability to perform cell lysis on Mycobacterium and Staphylococcus species. Some important notes include:
The amount of amplifiable DNA released by AudioLyse was approximately half that released by OmniLyse
AudioLyse was less efficient at Lysing than both OmniLyse and the Mini-BeadBeater which was used as a gold-standard in this experiment
OmniLyse was difficult to multiplex (since a single pair of hands can only process one sample roughly every 2 minutes)
AudioLyse does not require user-intervention during the lysis operation and so maybe easier to integrate into point-of-care devices.
There were indicators that some RNA was destructed in the AudioLyse (as well as for the BeadBeater) which can be explained by the absorption of RNA to the silica beads.
Concepts for Lab-on-a-chip Integration
These papers discuss devices that can integrate bead beating lysis and detection functionality for a lab-on-a-chip approach. The methods mentioned can help us decide on a feasible prototype if we decide to go with bead beating.
Berasaluce et al., 2015 describes a bead beating-based miniaturized cell lysis device that works in continuous flow allowing the analysis of large volume of samples without previous treatment. A permanent magnet and silica beads are placed inside a lysis chamber manufactured out of COP polymer and a rotating magnet creates an external magnetic field that causes the motion of beads within the chamber.
Pros:
Allows integration of a monolithic sample to answer system (OmniLyse is an off-chip device and a BeadBeater requires tubes and pertinent connections for sample transport
Suitable for applications which require large sample volumes such as water-based bacteria
COP material for body is biocompatible and compatible with commercial fabrication techniques such as injection-molding
Continuous flow lysis minimizes time consumption
Cons:
Low lysis efficiency relative to off-chip bead beating
The optimum configuration necessitates a compromise between time consumption and efficiency (60 µL/min lysis flow rate yielding 43% lysis efficiency relative to off chip bead beating)
The device is designed as follows:
Beckers et al, 2011 describe a device based on a free-mounted small chamber (typical volume = 100 microliters) made of a flexible material whose bottom is connected to a vibrating mechanical actuator. The actuator operates at one or two resonance frequencies and causes the vibration/deformation of the chamber resulting in bead collisions and/or inter-bead shear-flow. Cells are dispersed in water or PBS before introduction into the lysis chamber and cell rupture takes place when the kinetic energy of the colliding beads exceeds the elastic energy stored in the cell.
Note: The analysis included in this paper shows that the configuration is experimentally achievable but not yet optimal.
Notes on Usage of OmniLyse/AudioLyse
Bead Beating + Sonication + Atomization Device
Lim et al., 2015 describe a device made by commercial air humidifier parts that is capable of lysing 250uL of Bacillus subtilis to get a lysis efficiency of 66% in 30s. This device combines bead-beating by using 0.5mm- 1mm glass beads, sonication by using a piezoelectric disc (PZT) + perforated steel diaphragm + driver circuit from a humidifier (The driver circuit itself could be operated at 5 V and was powered via USB. In other words, it could either be powered via a laptop computer or a mobile device charger. The overall power consumption was 2 W). The overall cost of the device was less that 30 USD.
A big con of the device is that it cannot be reused after one round of lysis as it is contaminated. I am not sure if it can be cleaned in some way but the authors do not mention any such thing.Diagram of the deviceDevice setup
Electrical Lysis
Applying an electric field on a sample of cells can cause cell membrane disruption. The paper embeded below reports a throughput of 600 μl/min with a 90% cell lysis efficiency. This one also uses a similar complex manufacturing process to fabricate microelectrodes.
Some electrical lysis papers use lysozyme in addition to electrical lysis to increase lysis efficiency. A possible decision for us would be to use chitinase instead of lysozyme.
Groups have used electrical lysis by fabricating micro electrodes to generate very high potentials for cell lysis using voltages under 10 V:
Lu et al., 2006 fabricated a MEMS system with 3-D micro-electrodes [using a complex manufacturing process] to lyse leukocytes a 10 V, 100 µs pulse, achieving 30% lysis.
Lu et al., 2005 fabricated a microchip with a micro-electrode array [using a similar complex manufacturing process] to lyse HT-29 cells using AC at 10 kHz, ∼8.5 V, with 74% lysis.
Jiang et al., 2016 fabricated a microchip with a micro-electrode array [using a another manufacturing process] to lyse PK15 cells using a 6.8 V square wave pulse with a pulse width of 1 ms and period of 2.5 ms to get 50% lysis.
Gabardo et al., 2015 fabricated a microchip with a gold-based ($$$) micro-electrodes [using an easier manufacturing process] to lyse E. Coli cells using a 4 V square wave pulse with a pulse width of 500 ms at a 0.67 Hz for 30 s (20 pulses total) to get 95% lysis.
Rapid Nucleic Acid Extraction Through Magnetic Silica Beads
Here, Yamaguchi et al. 2016 describes an automated device for nucleic acid extraction via magnetic silica beads. The whole system consists of a mechanical control system and a disposable cassette. As shown in the figure, the disposable cassette consists of a bottle, containing the sample, the capillary tube, containing the extraction system, and the chamber, where the extracted nucleic acids is collected. The paper reports an automated extraction time of 3 minutes.
Two parameters that can affect nucleic acid extraction efficiency:
An integrated microfluidic device for DNA purification and PCR amplification of STR fragments
Seems to be the simplest method of silica solid phase extraction in a microfluidic chip
This chip was manufactured to have two functions: purify DNA and perform PCR. Each function will happen in two separate regions of the chip: the silica phase extraction (SPE) domain (Where DNA purification occurs) and the PCR domain (Where PCR happens).
How the chip was manufactured:
Taken directly from the paper, "Borofloat glass (Telic Company, Valencia, CA) bottom plates for the microdevices were fabricated using standard photolithographic techniques as described previously[37]. The design used for the integration of SPE-PCR is shown inFig. 1and all microchips were etched to a depth of 200mm. The mask consisted of the SPEchannel 15 mm0.10 mm (all dimensions given as length width) with a weir 2 mm from the exit of the channel(the gap between the top of the weir and cover plate was approximately 15mm). The side channel, used for the introduction of PCR reagents, was 11.1 mm0.025 mm, and the PCR channel was 9.1 mm0.025 mm. An ellipse was placed in the middle of the PCR channel with the dimensions 3.0 mm0.75 mm."
The solid phase was then created by packing silica beads, 5–30mmdiameter, suspended in ddH2O, into the channel against the weirusing vacuum.
Total time for purification: ~16 minutes and 30-45 seconds
Steps of the SPE domain:
The lysed sample in 6 M GuHCl, was loaded onto the bed for 6 min (150 nL semen loaded) (6 minutes)
Proteins and PCR inhibitors were removed from the SPE bed by passing wash solution (2-propanol/water, 80/20 (v/v)), through the SPE domain for 10 min. During loading and wash steps, ddH2O was rinsed through the side channel of the microdevice. (10 minutes)
DNA was eluted from the SPE domain with water and/or reaction buffer as detailed, as PCR master mix was simultaneously perfused through the side channel. (30-45 seconds)
Yamaguchi et al., 2015 describe an automated device consisting of a mechanical control system and a disposable cassette for the extraction of nucleic acids in around 3 minutes.
The mechanical control system consists of a motor, a magnetic force application unit, and a pair of permanent magnets. The magnetic silica beads are moved in the longitudinal direction by the motor and both the permanent magnets and the magnetic force application units vibrate the magnetic beads in a zigzag motion. According to the authors, this system efficiently washed out various inhibitors that could prevent PCR analysis, and dramatically shortened the extraction time.
Note that this device essentially focuses on isolation and purification more than extraction.
Berry et al., 2016 introduce a device called AirJump for the extraction of viral RNA with sufficient repeatability and sufficient purity to quantitate with qRT-PCR.
The AirJump apparatus consists of a base with alignment posts, a standard 96-well plate, elution wells configured as a strip or plate, and a magnet holder. The 96 well-plate is placed on the base and each well is loaded with with 420 μL of solution containing sample, buffer, and PMPs. Then, the alignment posts are used to position the sample and elution wells face to face and a holder containing an array of magnets is placed above the elution well. The PMP -bound analyte is then 'jumped' through a liquid/air interface and air gap of (~1mm).
This process takes 0.5-2 minutes for PMP transfer and once done, the AirJump apparatus is disassembled and a multichannel pipet is used to resuspend the PMP-bound analytes (now located in the elution wells) in elution buffer. In this paper, the PCR analysis showed that the RNA extracted through AirJump showed a high degree of accuracy and precision. This separation technique is an example of ESP (exclusion-based sample preparation) which involves drawing the analyte magnetically through an interface (e.g., aqueous/air, aqueous/oil) to separate it from the unbound material. Traditionally, PMP-based techniques use multiple wash steps with the buffer to remove the background from the bound fraction.
To do: extracting DNA from PMP
Notes:
percentage of the PMPs that was recovered ranged from 77% to 87%, and recovery became higher as PMP concentration was increased
Based on the author's characterization experiments, with some process optimization, AirJump can achieve very high purity (less than 0.5% carryover) and low PMP loss (less than 15% total loss)
AirJump carryover is highest with high surface tension buffers and decreases as surface tension is reduced (so AirJump performance can be enhanced by adding detergents to the sample) . Detergents are commonly found in lysis buffers used in protein and nucleic acid extractions)
AirJump recovers slightly more protein than the washing protocol (72% recovery vs 56% recovery
Advantages:
Since ESP does not dilute or discard the original sample during isolation, this material can be “resampled” multiple times to extract different analytes sequentially
Due to "air-jumping", purification efficiency is higher than tube-based method due to lavck of surface-based contaminants.
PMPs are not drawn along a surface, mitigates friction and adsorption
Due to well plate, extractions can be performed in parallel (96 extractions simultaneously
AirJump removes approximately 2-fold more background compared to the wash
Disadvantages:
Surface tension and concentration of the buffer impacts AirJump performance since the size of the “jumping” PMP aggregates will change
"(A) Standard washing paradigm where PMP-bound analyte is magnetically pulled to the side of a tube and washed several times. (B) Mechanisms of carryover with standard method including tube residue and PMP-trapped material. Photo of AirJump components in disassembled (C) and assembled (D) states. (E) Schematic of AirJump operation; an elution plate is inverted and placed above a sample plate loaded with target analyte (blue) and nontarget material (green) (1). Upon application of a magnet, PMP-bound analyte “jumps” across the air gap and is deposited in the elution plate (2)."
Microfluidic Chip/LOC Approaches:
Lee et al., 2006 describe a Laser-Irradiated Magnetic Bead System (LIMBS) for cell lysis and DNA isolation.
Microchips with a chip size of 7.5 mm × 15 mm and 10 µl sample volume were fabricated using silicon, glass, polycarbonate film, and double-coated tape in order to measure the temperature of solution and perform laser-induced sample preparation. Pathogens and magnetic beads are loaded into these disposable microchips which are then placed into a chip guide module. For lysis, a high power laser beam at 808 nm was applied to the microchip concurrently with vibration provided by a portable motor. This wavelength is selected such that most of the laser beam is transmitted through the water and reaches the magnetic beads with only a small amount of absorption. Cell lysis is most likely caused by the combination of the heat shock and the mechanical shock delivered by the magnetic beads, highly heated magnetic beads bump against the cells, transfer heat and disrupt the cell walls and membranes. In this study, Gram-positive bacterial cells (Streptococcus mutans, Staphylococcus epidermidis) as well as HBV mixed with human serum were easily disrupted within 40 seconds in the microchip.
Notes:
DNA extraction and real-time detection of pathogens in a single chamber of a microchip happened within 32 minutes.
Can use the same microchip for sample preparation and real-time pathogen detection in a PCR machine without changing the solution
The survival rate of cells decreased (lysis efficiency increased) with increased laser power and exposure time
Sample preparation by LIMBS showed higher efficiency of DNA release from the same number of cells than other conventional cell lysis methods (shown in figure 6)
"(a) Schematic diagram of a microchip for temperature measurement. (b) Schematic diagram of a hand-held type LIMBS using small laser diode and microchip holder with vibration motor. (c) Image of hand-held type sample preparation device (58.3 × 57.9 × 37.0 mm, 148 g) with a small laser diode."
Liu et al., 2009 present a microfluidic device for rapid RNA extraction without any manual operation or the addition of unstable inhibitor substances.
The RNA is released from the cells by a lysis buffer solution and trapped onto positively-charged magnetic beads (due to the pH being lower than 6.0). Meanwhile, protein and contaminants are not bound to the beads and can be simply washed away using micropumps. RNA is then purified and collected by an on-chip magnetic bio-separator consisting of twisted microcoils which are supplied with a DC current to attract the magnetic beads onto the surface of the microchip. An elution buffer is then added to the RNA bound on the magnetic beads for purification and finally, the purified RNA can then be reverse transcripted into cDNA using a built-in micro-heater.
IFAST - Berry et al., 2014 describe a device for RNA extraction using immiscible filtration assisted by surface tension (IFAST)
Device:
Produced via embossing of wax using a stamping process (inexpensive and environmentally friendly when disposed of)
Uses surface tension to position aqueous and oil phases side by side, such that nucleic acid (or another analyte) can be bound to a PMP in a sample and drawn through an oil phase into an elution buffer
The device is placed on a flat surface then loaded with the aqueous and immiscible oil phases
The sample with the PMPs is loaded into the input well (100-500ul depending on the size of the device used)
Magnets are then slid under the device (by hand at a speed of about 1-2mm per second) to move the sample from the input to the output well
"Figure 1. Diagram of the IFAST design. Connected wells in the center are used for analyte extraction, whereas wells on either side are for fabrication purposes only (A); schematic of wax IFAST fabrication (B); image illustrating the difference between using a single oil barrier (left) and two oil barriers (right) (C); schematic of IFAST sample extraction procedure (D)."
Notes:
To quantify viral RNA extraction, RT-qPCR was performed on the IFAST-extracted RNA. After IFAST operation, the eluent containing the PMP-bound RNA was extracted via pipette and mixed with an equal volume of 2× reverse transcription master mix (High Capacity cDNA Master Mix; Life Technologies, Carlsbad, CA).
Small magnetic cubes (BX333-N52; K&J Magnetics, Pipersville, PA) were used for these experiments
By changing the mold, the device can be manufactured with different number of wells and varying volumes (Input volumes used were: 100 μL, 200 μL, 350 μL, and 500 μL)
Using FC-40 oil for the oil phaseresulted in the lowest percent contaminant carryover at 1.7% ± 0.2% and 0.7% ± 0.3% for one and two oil barriers, respectively
Note that aqueous phases would often connect on addition of mineral or silicone oil ("the aqueous sample wells were observed to creep underneath the adjacent oil phase, eventually connecting to the next aqueous well, resulting in mixing of the well contents. This failure mode was especially evident when devices were picked up for operation, likely because of the slight tilt imparted to the device. FC-40 oil demonstrated superior pinning stability, preventing excessive carryover of sample into the output elution well, and thus was selected for subsequent trials.")
If the wax surface wasn't smooth (due to the kinetics between the mold and the wax), some PMPs would get trapped in the wrinkles of the device. (This was especially true for larger input wells which had higher surface area)
The authors were able to achieve consistently smooth devices with an automated, Peltier-heated embossing system (but this is more expensive and complex than what they used when the devices weren't completely smooth)
The authors anticipate the 500ul version of the device to have the highest sensitivity despite the fact that it generated a higher CT value.
Sur et al., 2010 describe a method for the purification nucleic acids using paramagnetic beads.
Theory
"Extraction and purification of nucleic acids from complex biological samples for PCR are critical steps because inhibitors must be removed that can affect reaction efficiency and the accuracy of results. This preanalytical processing generally involves capturing nucleic acids on microparticles that are then washed with a series of buffers to desorb and dilute out interfering substances."
In this paper, the authors propose a method for purification that replaces these multiple wash steps with a single pass of Paramagnetic particles (PMPs) through an immiscible hydrophobic liquid. Only two aqueous solutions are required: a lysis buffer, in which nucleic acids are captured on PMPs, and an elution buffer, in which they are released for amplification.
Process
"The PMPs containing the nucleic acids are magnetically transported through a channel containing liquid wax that connects the lysis chamber to the elution chamber in a specially designed cartridge. Transporting PMPs through the immiscible phase yielded DNA and RNA as pure as that obtained after extensive wash steps required by comparable purification methods."
Typically, PMP-based systems process samples in a single well by repeatedly pelleting PMPs, aspirating the liquid, and adding wash solution. NAs are released from the cells and bound to PMPs after a lysis buffer is added, multiple wash steps then remove amplification inhibitors, and NAs are eluted from the PMPs to yield a purified and concentrated sample.
Instead, the author's strategy for this method is to transfer the PMPs between wells in a specially designed cartridge with an externally applied magnetic field eliminating all contact between the processing system and the sample. The wells are connected with a hydrophobic liquid through which PMPs are transported and this liquid acts as an immiscible phase filter between the lysis chamber and the elution chamber. This allows for a reduction in the number of steps and time required for purification while also simplifying the instrumentation and reducing the number of consumables.
Advantages of using this method with PMPs:
NAs can be isolated from crude sample materials
Wide ranges of sample volumes can be accommodated
Large batches of samples can be processed without centrifugation
simple and reliable way to filter PCR inhibitors and eliminate wash steps
A note about this method: keep in mind that the authors mainly applied this immiscible-phase process to targets in whole blood, plasma, and urine. Essentially, the paper focuses on complex bio samples like blood, biopsied tissue, cultured cells and food to name a few but the PMP idea might be useful for us to explore
Note that the authors also mention that a magnetic mixer/mover was built for uniform performance and uses a stepper motor and gears which might not exactly be easy
Silica Matrices
Theory
Using silica matrices is a well known method for purifying DNA by exploiting its negatively charged backbone. Positively-charged silica acts as a selective filter to DNA by tightly binding to it. Sodium cations in the solution can break the hydrogen bonds between the hydrogen in water and the negatively charged oxygen ions in silica under high salt conditions (pH ≤ 7). The DNA is tightly bound, and extensive washing removes all contaminations. The purified DNA molecules can be eluted under low ionic strength (pH ≥ 7) later by using TE buffer or distilled water. - Tan. 2009
Branch et al. 2017 employs a sonication technique for lysis and a silica matrix medium for purification, all done on a microfluidic chip.
They fabricated a sonication platform using gold electrodes and piezoelectric substrates which was able to lyse 50% of 50uL or E.Coli cells in 20s
They also tested a few DNA extraction methods on this lysate
Sol-Gel Packed Microchannels: The lysate was flown through a silica beads + sol-gel filled channel and then washing and elution (using TE buffer) was done to extract the DNA from the gel to get a 40% efficiency.
ChargeSwitch® Magnetic beads: The DNA in lysate was extracted using magnetic beads packed inside the channel and then changing the pH to bind, wash, and elute the DNA to get 38% efficiency and 58.9 % efficiency with acoustic mixing.
Naflon-based Cartridge: The lysate was passed through naflon coated electrodes which bound the DNA in the nalfon coating and then after washing, this DNA was eluted by changing the polarity of the electrodes, the efficiency was 69%.
Zhao et al's universal DNA extracting syringe
Published last year on nature, Zhao et al. 2019 develops "a simple, universal protocol for use in nucleic acid testing-based pathogen diagnostics, which requires only hand-powered sample preparation, including the processes of pathogen enrichment and nucleic acid isolation"
There are two stages:
Pathogen Enrichment
Nucleic Acid Extraction/Purification
This system uses a syringe filter containing amine-functionalized diatomaceous earth (ADE) and homobifunctional imidoesters (HI) that traps the pathogens. Here, ADE functions as a means for pathogen enrichment by trapping them in the filter. More specifically, it achieves this goal due to the positively charged ADE absorbing negatively charged pathogens. HIs, on the other hand, enhances pathogen enrichment by reacting with the amine groups of ADE to form even more positively charged groups. HIs have another purpose: they can reversibly crosslink amine groups on nucleic acids. Furthermore, this reaction can be reversed (un-crosslink) by changing the pH, ultimately allowing nucleic acids to be eluted out of the syringe for collection.
Figure. Diagram illustrating the whole process
Whatman FTA Card
Whatman FTA card is used prior to DNA extraction step including the official instruction guide.
This facilitate collection, transport, purification, and long-term, room-temperature storage of nucleic acid.
The fundamental of FTA™ cards is that it lyses the eukaryotic cells that are in contact with the cards. After lysis the proprietary chemical content of the card protects the DNA from the environmental and enzymatic damages.
DNA remains bound to the matrix of FTA cards, which can then be used directly for downstream applications. With FTA Elute cards, DNA will be eluted into water after purification. The eluted DNA can be used for quantitative DNA analysis.
Whatman FTA filter matrices are fibrous cards impregnated with chelators and denaturants that lyse and inactivate most microorganisms.
lysis of cells, designed to fix and store DNA directly from tissue while preventing growth of bacteria.
Released large nucleic acids become physically trapped within the fibres of the FTA matrix and are preserved intact, while cellular debris can be rapidly removed by simple washes of the inoculated card
Individual filter punches were removed from each card using biopsy punches.
Filter punches were then washed with FTA wash buffers, and added directly to PCRs. Or nucleic acid extraction step follows.
Usage
Ward et al., 2018 uses Whatman FTA card as a sample collection swab from amphibian's skin.
PCR success rate of 66% (FTA card with washing) and 78% (FTA® card punches from Qiagen-based DNA extraction)
Require five times washing with 2 different buffers for 3-5 min or used with DNA extraction kit (Qiagen DNeasy Blood & Tissue kit)
In buffer method, the dried punches are added to PCR mastermix
Qiagen DNeasy Blood & Tissue kit is not feasible: require overnight incubation, centrifuge
Rathore et al., 2019 uses Whatman FTA card for sample collection (+following RPA reaction)
42.0 mm-diameter discs were punched from each FTA card using a Harris micropunch tool and washed with an FTA purification reagent followed by another washing using TE buffer twice as and when required. The discs were then used directly as the DNA template for the positive (with template) RPA assay.
Limitation:
Possibility of any inhibitory effect due to the composition of the FTA matrix on the RPA reaction was investigated. Any remaining components of the FTA wash buffer might inhibit and/or decrease the yield of the RPA reaction. Hence, additional wash of the FTA card with TE buffer was observed to be necessary for a successful RPA reaction.
Require multiple FTA disks to supply enough template (low yield for each disc)
The need for a sensitive system to discriminate true-positive results from false-positive and/or negative results in case of a low template load in the reaction
Using this method, additional purification step on RPA reaction was required.
No info about extraction time given in the paper
M. Borman et al., 2010 improves Whatman FTA card to be cost effective, rapid, reduces potential cross-contamination between sequential filters
3-4 freeze thaw cycles coupled with vortex mixing in presence of glass beads of sample
then add the prepared solution to FTA filter punch
inoculated filters are air-dried for 5-10 min and then washed
FTA card is used for extraction and purification of DNA in this POC device
DNA was extracted by employing a small FTA card (3 mm in diameter) containing a mixture of strong buffers and chemicals (FTA purification reagent and TE buffer for 5 min at room temperature, separately)
All washing solutions were discarded using pipette
Device illustration and work flow:
Extraction time: 30min
From these results, it could be concluded that it was likely difficult to extract DNA from Gram-positive bacteria (S. aureus) as compared with Gram-negative bacteria (Salmonella spp. and E. coli O157:H7) by using the FTA card
The Limit Of Detection of bacteria (E. coli) was approximately 3 × 101 CFU/sample
Owing to the use of 3 μL of bacteria sample *S. aureus) for each reaction, the LOD was approximately 3.0 × 102 CFU/sample
Detection limit may differ for fungal sample
Trinh et al., 2020 Same extraction method, but different microdevice feature
FTA Elute card
Hashimoto et al., 2019 suggests that FTA Elute card yield the best template DNA for PCR over FTA card and 3MM Chr papers
This protocol follows the official protocol by GE Healthcare Life Sciences
DNA was extracted from DBS containing blood on FTA Elute cards using the QIAmp DNA Micro Kit.
DBS was rinsed trice with water by pulse vortexing
Then, water was added to the DBS and the card was incubated at 95 °C for 30min
5-10μL of Eluted template DNA is added to PCR reaction mix
LoD was 1.6 parasites/μL blood for the FTA Elute Card
not feasible
Limitation
Zhao et al. 2019 reports FTA card-based assays to face difficulties when under low concentrations (on order of femtomolar or nanomolar) of analytes or small volumes (1–1000 µL). This would make downstream detection to be much less efficient as the FTA paper would serve as a bottleneck for the "specificity" of the product.
Volume and concentration depend on nucleic acid extraction procedures and results.
For nucleic acid detection process, it is preferred to test on small volume considering the cost of primers and other reagents in the downstream reaction. iGEM 2019's project, we consider the volume of nucleic acid to be 5µL.
2. Cards need to be dried for at least 2 h at room temperature before sample collection and 3-5 min after sample collection (M. Borman et al., 2010).
3. Detection limit of the FTA card for both RNA (105 CFU) and DNA (103 CFU) was less sensitive than that of our assay (our filter containing the ADE and DMS system) (Zhao et al. 2019)
4. PCR success rate of 66% using washing buffer (M. Borman et al., 2010): FTA card and punches is used as extraction and purification method.
5. Many protocols require vortexing and incubation at 95 °C
6. Danger of cross-contamination from filter to filter when filter punches are removed
Other Methods
Purification via Chitosan membrane
In Byrnes et al. 2015, they develop a DNA purification system in lateral flow format via a porous chitosan membrane. Chitosan, a positively charged derivative of chitin, is used as a membrane that would allow any material to pass through, except negatively charged DNA. In this study, Byrnes et al. tests the chitosan technique in two different porous membranes: nitrocellulose and glass fiber.
The capacity for DNA in nitrocellulose was 1.9 × 10^6 copies of DNA per μg of chitosan
The capacity for DNA in glass fiber was 9.9 × 10^6copies of DNA per μg of chitosan
This figure illustrates how DNA can be eluted out of the membrane by simply altering pH. In solutions with a pH less than 6.3, protonated primary amines on the chitosan and allow DNA binding. When the pH is brought above 6.3, the charges on the primary amines return to neutral and DNA is released.









https://www.golledge.com/products/saw-resonators-from-golledge-electronics/c-26/c-86