Figure 5. Initial conformation of Gelidibacter algens candidate protein at 0 ns of molecular dynamics production.
Team:ULaval/Model
PROJECT MODELLING
Identifying a candidate
dextranase for our project
Once we determined our project for the 2020-2021 iGEM competition would be expressing a dextranase to treat ropy maple syrup, we set out to look for our candidate enzyme. We identified several conditions that we should consider when selecting our candidate dextranase:
- Ropy maple syrup is stored at room temperatures (~10-15°C): Our enzyme would need to retain a high activity at those temperatures, whereas most dextranases have optimal temperatures around 50 - 60° C (Khalikova et al., 2005; Ren et al., 2019).
- Ropy maple syrup has a high sugar concentration and viscosity: These inherent conditions of ropy maple syrup can reduce the catalytic efficiency of our enzyme. In particular, dextranases have been shown to have a lower activity at high sugar concentrations (Bashari et al., 2013).
In view of these requirements, we decided to use a bioinformatics approach to select a candidate enzyme from a psychrophile organism. Using several techniques, we aimed to validate the conservation of active site residues to make sure our protein was indeed a dextranase.
Selecting a candidate dextranase
We surveyed the literature and identified that there are two distinct families of dextranases (Larsson et al., 2003; Ren et al., 2019; Suzuki et al., 2012). As shown in Figure 1, we selected three reference dextranases whose sequences and structures were available and used BLASTP (Camacho et al., 2009) against the non-redundant database to identify homologous sequences for each of them. Using those lists of homologous sequences, we used references on the taxonomy of psychrophiles (Nogi, 2011) to identify dextranases expressed by these organisms. Based on this analysis, we identified two candidate dextranases from the psychrophilic organisms Gelidibacter algens and Aureobasidium subglaciale. We realigned these two candidate sequences to our reference dextranases using MAFFT (Katoh et al., 2002; Katoh & Standley, 2013) and confirmed that key residues for their catalytic activity were conserved in the active site (Figure 2). However, we observed that A. subglaciale is an eukaryote, which could complicate the expression of its dextranase in our chassis of choice, Escherichia coli, due to the lack of posttranslational modifications in prokaryotes. Finally, we decided to move forward with the dextranase from Gelidibacter algens, which we submitted to the iGEM registry as part BBa_K3493000.

Figure 1. Scheme of the process used to select candidate dextranases from psychrophilic organisms.
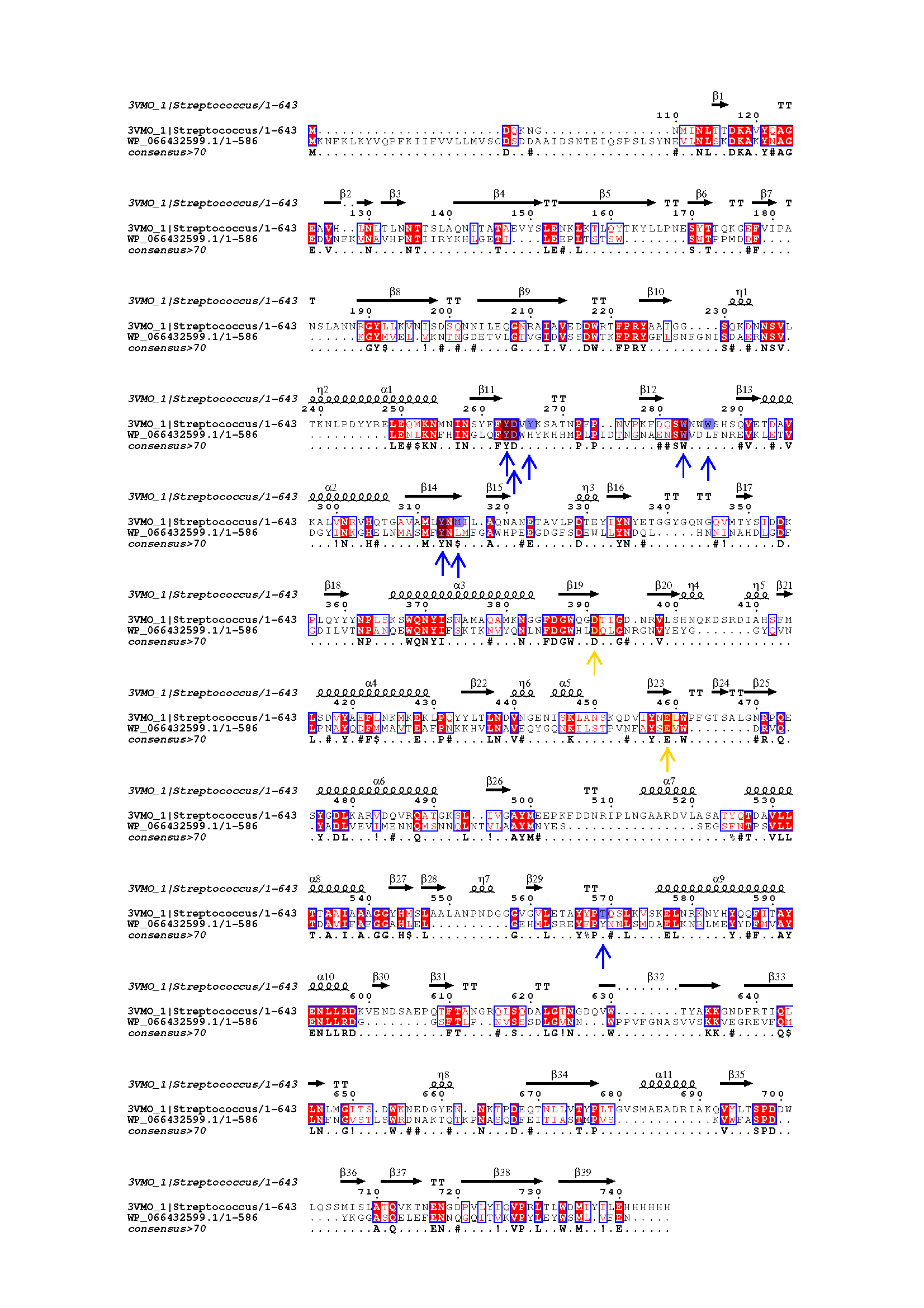
Figure 2. Sequence alignments of reference Streptococcus mutans and candidate Gelidibacter algens dextranases. Yellow arrows show catalytic residues, blue arrows show other important reference dextranase residues.
Structural modeling and
validation of the active site
validation of the active site
While the key catalytic residues were conserved in our candidate dextranase, we also observed that sequence identity between the candidate and the reference was modest at 24.61%. Thus, sequence identity is below the threshold at which proteins have been generally observed to have the same fold, although it is still higher than for random proteins (Addou et al., 2009; Todd et al., 2001). Still, we decided to continue our approach with this candidate due to the lack of characterized psychrophilic dextranases within the family 66 of such an enzyme.
We used homology modelling servers I-TASSER (Zhang, 2008) and SWISS-MODEL (Waterhouse et al., 2018) to gain further insight into the structure of the G. algens dextranase. These servers infer the structure of a query protein based on a series of homologous proteins for which the structure has been solved. We superposed the structure provided by the servers to our reference dextranase from Streptococcus mutans and compared the positions of the active site residues. Indeed, we observed that the critical active site residues have a similar configuration in the active site, which increased our confidence in our protein actually being a dextranase (Figure 3).
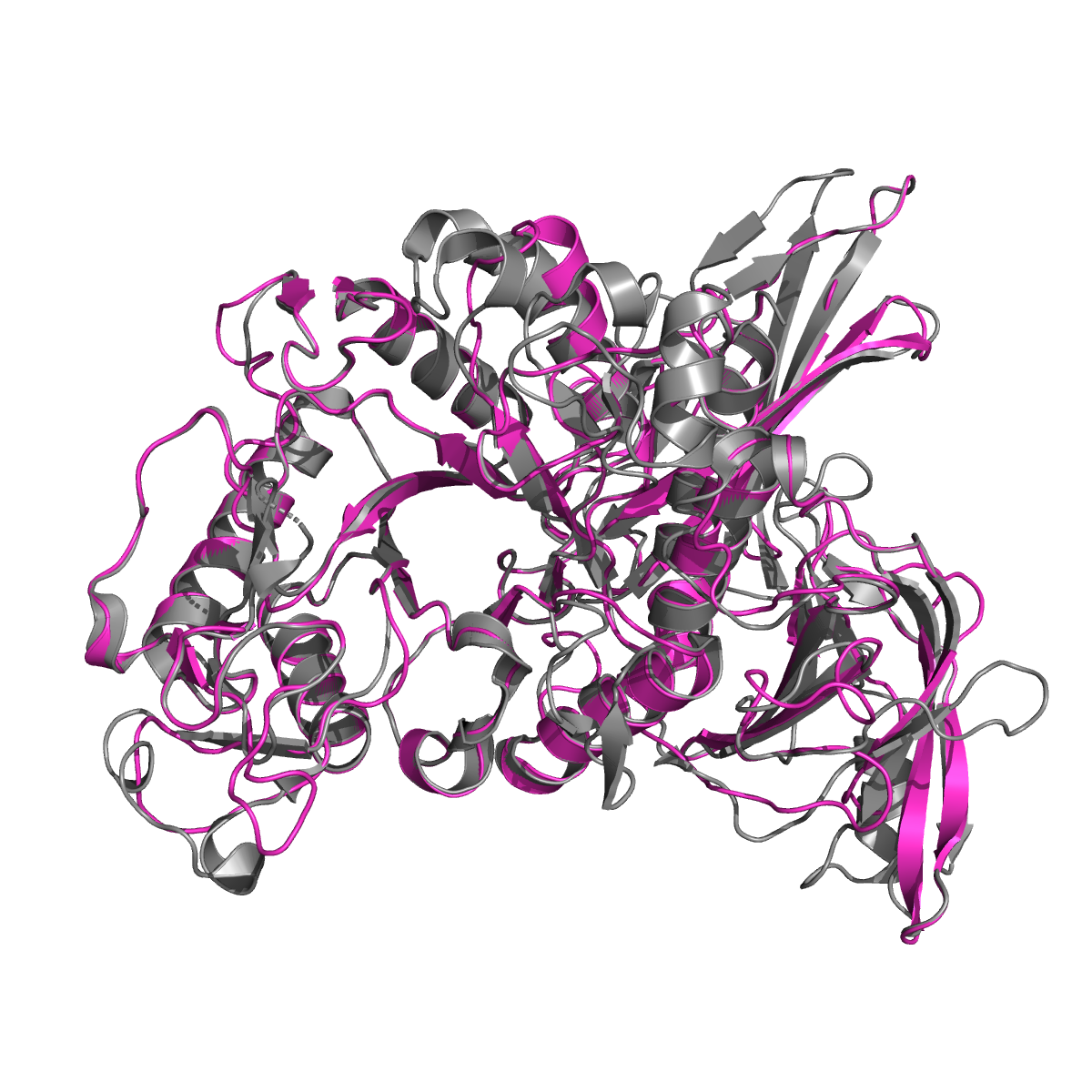
Figure 3. SWISS-MODEL alignments of the structures of Streptococcus mutans (gray) and Gelidibacter algens (pink) with a RMSD of 0.247.
Docking and molecular dynamics
Taking our analysis further, we performed molecular dynamics on our candidate dextranase from G. algens. Our main goal was to determine its overall structure conservation through time, specifically the active site. In order to do so, we prepared a molecular dynamics system of the dextranase structure provided by SWISS-MODEL (as the I-TASSER structure showed striking similarity) with CHARMM-GUI Solution Builder (Jo et al., 2008; Lee et al., 2016). The protein was simulated at 297.15 K in a 0.15 M NaCl water solvent. Due to time constraints, the results were taken from a trajectory of 76 ns, but the complete analysis (planned for the iGEM 2021 edition) will include at least 200 ns of trajectory in order to study possible slower, more functionally important protein movements. An important part of molecular dynamics done on our candidate dextranase was the parameterization of the simulation conditions. Figure 4 shows stable temperature values after equilibrating the system and throughout the production trajectory. This test helps us make sure our simulation reflects actual molecular behaviors instead of unwanted artifacts. This parametrization will be reused for subsequent simulations in the following 2021 iGEM edition.
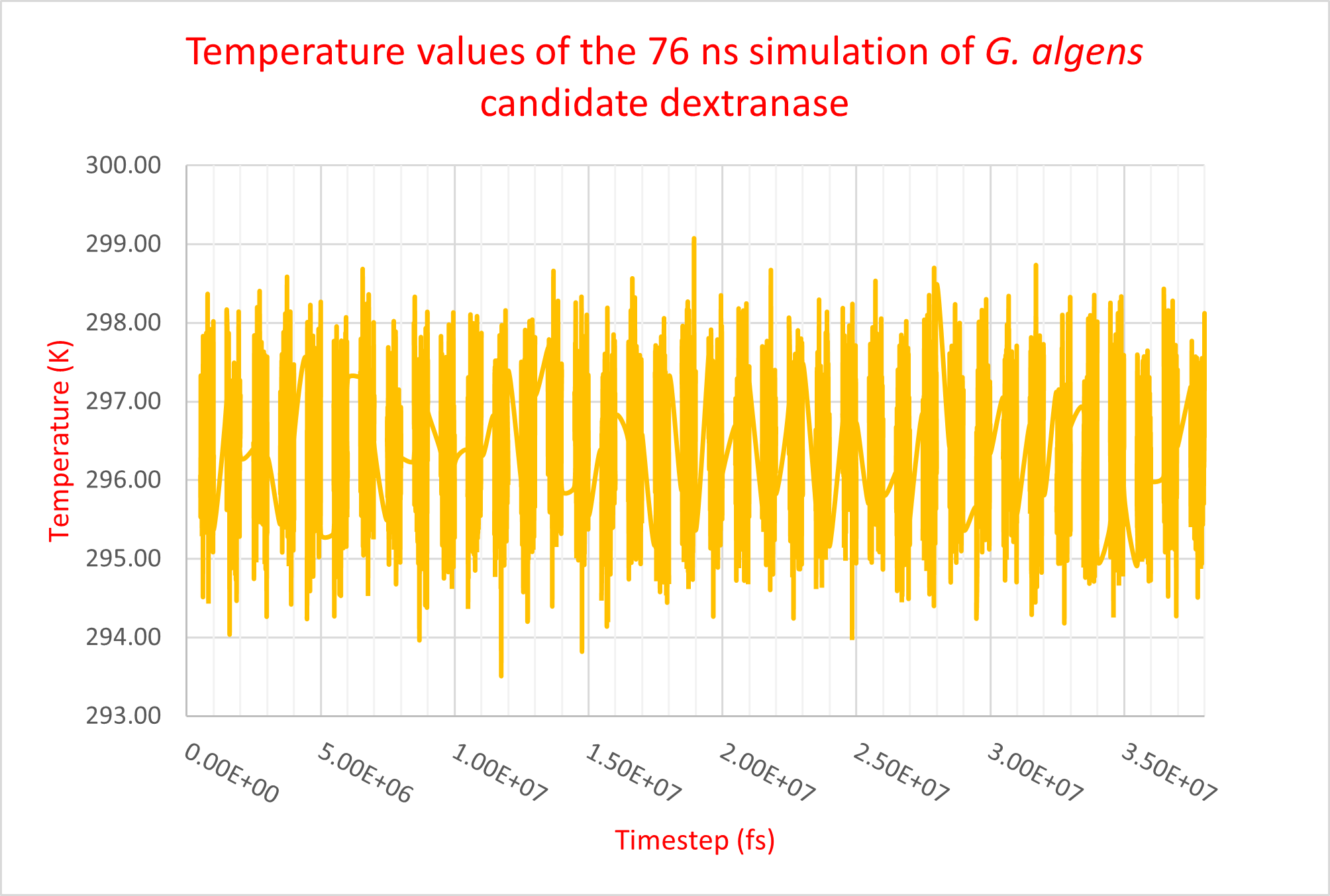
Figure 4. Temperature values of the 76ns molecular dynamics trajectory of G. algens candidate dextranase. Each data separated section shows a 2 ns long trajectory.
|
1 / 7

2 / 7

Figure 6. Conformation of Gelidibacter algens candidate protein at 19 ns of molecular dynamics production. 3 / 7

Figure 7. Conformation of Gelidibacter algens candidate protein at 38 ns of molecular dynamics production 4 / 7

Figure 8. Conformation of Gelidibacter algens candidate protein at 57 ns of molecular dynamics production. 5 / 7

Figure 9. Final conformation of Gelidibacter algens candidate protein at 76 ns of molecular dynamics production. 6 / 7

Figure 10. Structural alignment of the initial (0 ns, pink) and final (76 ns, salmon) structures of molecular dynamics production. The conserved non-catalytic residues of the two structures are shown in blue (initial) and cyan (final) while the conserved catalytic residues are shown in yellow (initial) and green (final). 7 / 7

Figure 11. Active site of structural alignment of the initial (0 ns, pink) and final (76 ns, salmon) structures of molecular dynamics production. The conserved non-catalytic residues of the two structures are shown in blue (initial) and cyan (final) while the conserved catalytic residues are shown in yellow (initial) and green (final). |
In order to study structural changes in our candidate dextranase, we sampled structures at five different timestamps : 0 ns (initial production structure), 19 ns, 38 ns, 57 ns and 76 ns (final structure). Every 19 ns segment represents a fourth of the production time length. Between each structure, there are minimal differences in α helices and β sheets positions, but substantial loop divergence. The comparison between the initial (t = 0 ns, pink) and final (t = 76 ns, salmon) structures shows a RMSD of 3.627 hinting at modest structural identity. Figure 10 and 11 show poor conformational conservation of non-catalytic conserved residues (blue for initial structure, cyan for final structure). This can be explained by their positioning in or near loops whose conformations vary significatively throughout the simulation. On the other hand, conserved catalytic residues (in yellow) stay in the same relative area of the active site, showing evidence of a stable dextran-friendly interaction environment. |
|
1 / 7
Figure 5. Initial conformation of Gelidibacter algens candidate protein at 0 ns of molecular dynamics production. 2 / 7
Figure 6. Conformation of Gelidibacter algens candidate protein at 19 ns of molecular dynamics production. 3 / 7
Figure 7. Conformation of Gelidibacter algens candidate protein at 38 ns of molecular dynamics production 4 / 7
Figure 8. Conformation of Gelidibacter algens candidate protein at 57 ns of molecular dynamics production. 5 / 7
Figure 9. Final conformation of Gelidibacter algens candidate protein at 76 ns of molecular dynamics production. 6 / 7
Figure 10. Structural alignment of the initial (0 ns, pink) and final (76 ns, salmon) structures of molecular dynamics production. The conserved non-catalytic residues of the two structures are shown in blue (initial) and cyan (final) while the conserved catalytic residues are shown in yellow (initial) and green (final). 7 / 7
Figure 11. Active site of structural alignment of the initial (0 ns, pink) and final (76 ns, salmon) structures of molecular dynamics production. The conserved non-catalytic residues of the two structures are shown in blue (initial) and cyan (final) while the conserved catalytic residues are shown in yellow (initial) and green (final). |
In order to study structural changes in our candidate dextranase, we sampled structures at five different timestamps : 0 ns (initial production structure), 19 ns, 38 ns, 57 ns and 76 ns (final structure). Every 19 ns segment represents a fourth of the production time length. Between each structure, there are minimal differences in α helices and β sheets positions, but substantial loop divergence. The comparison between the initial (t = 0 ns, pink) and final (t = 76 ns, salmon) structures shows a RMSD of 3.627 hinting at modest structural identity. Figure 10 and 11 show poor conformational conservation of non-catalytic conserved residues (blue for initial structure, cyan for final structure). This can be explained by their positioning in or near loops whose conformations vary significatively throughout the simulation. On the other hand, conserved catalytic residues (in yellow) stay in the same relative area of the active site, showing evidence of a stable dextran-friendly interaction environment. |
For next year, we also plan to compare molecular dynamics results of our dextranase with other dextranases from mesophilic and thermophilic organisms. We aim to differentiate structural dependencies of similar enzymes that would explain the enzymes specificities according to their host’s temperature environment. Furthermore, we intend to study conserved residues RMSF between temperatures using methods similar to those previously described by Kovacic et al., (2016).
We also aim to further characterize the interactions of the G. algens dextranase with dextran. To do so, we have started evaluating docking options with Autodock Vina (Trott & Olson, 2010). This tool would allow us to predict the most favorable configuration of dextran in the enzyme’s active site, and in turn, compare it to the crystal structure of the S. mutans protein (Suzuki et al., 2012). We have generated some preliminary results but our docking pipeline still needs some refinement, which we will work on during the 2021 iGEM edition.
- Addou, S., Rentzsch, R., Lee, D., & Orengo, C. A. (2009). Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. Journal of Molecular Biology, 387(2), 416–430
- Bashari, M., Tounkara, F., Abdelhai, M. H., Lagnika, C., Xu, X., & Jin, Z. (2013). Impact of Dextranase on Sugar Manufacturing and its Kinetic on the Molecular Weights of Remaining Dextran. Sugar Tech, 15(1), 84–93.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics, 10, 421.
- Jo, S., Kim, T., Iyer, V. G., & Im, W. (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. Journal of Computational Chemistry, 29(11), 1859–1865.
- Katoh, K., Misawa, K., Kuma, K.-I., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066.
- Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780.
- Khalikova, E., Susi, P., & Korpela, T. (2005). Microbial dextran-hydrolyzing enzymes: fundamentals and applications. Microbiology and Molecular Biology Reviews: MMBR, 69(2), 306–325.
- Larsson, A. M., Andersson, R., Ståhlberg, J., Kenne, L., & Jones, T. A. (2003). Dextranase from Penicillium minioluteum: reaction course, crystal structure, and product complex. Structure, 11(9), 1111–1121.
- Lee, J., Cheng, X., Swails, J. M., Yeom, M. S., Eastman, P. K., Lemkul, J. A., Wei, S., Buckner, J., Jeong, J. C., Qi, Y., Jo, S., Pande, V. S., Case, D. A., Brooks, C. L., 3rd, MacKerell, A. D., Jr, Klauda, J. B., & Im, W. (2016). CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. Journal of Chemical Theory and Computation, 12(1), 405–413.
- Nogi, Y. (2011). Taxonomy of Psychrophiles. In K. Hirokoshi (Ed.), Extremophiles Handbook. Springer.
- Ren, W., Liu, L., Gu, L., Yan, W., Feng, Y. li, Dong, D., Wang, S., Lyu, M., & Wang, C. (2019). Crystal Structure of GH49 Dextranase from Arthrobacter oxidans KQ11: Identification of Catalytic Base and Improvement of Thermostability Using Semirational Design Based on B-Factors. In Journal of Agricultural and Food Chemistry (Vol. 67, Issue 15, pp. 4355–4366). https://doi.org/10.1021/acs.jafc.9b01290
- Suzuki, N., Kim, Y.-M., Fujimoto, Z., Momma, M., Okuyama, M., Mori, H., Funane, K., & Kimura, A. (2012). Structural elucidation of dextran degradation mechanism by streptococcus mutans dextranase belonging to glycoside hydrolase family 66. The Journal of Biological Chemistry, 287(24), 19916–19926.
- Todd, A. E., Orengo, C. A., & Thornton, J. M. (2001). Evolution of function in protein superfamilies, from a structural perspective. Journal of Molecular Biology, 307(4), 1113–1143.
- Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461.
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T. A. P., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303.
- Zhang, Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, 9, 40.