Team:Austin UTexas/Model
Contents
Modeling
![]()
To model our engineered T7 genomes, we are using the Pinetree stochastic gene simulator created by the Wilke Lab↗ at the University of Texas at Austin. This program is able to simulate the binding, transcription, and translation events that occur during gene expression at the molecular level using a Gillespie Stochastic Model Algorithm. For our purposes, we are using a model designed to replicate the environment of an Escherichia coli cell during the course of a T7 bacteriophage infection. This lets us view how the concentrations of ribosomes, polymerases, and various gene products change over the course of infection, as well as compare differences in expression between mutated T7 genomes.
T7 Modeling before PineTree
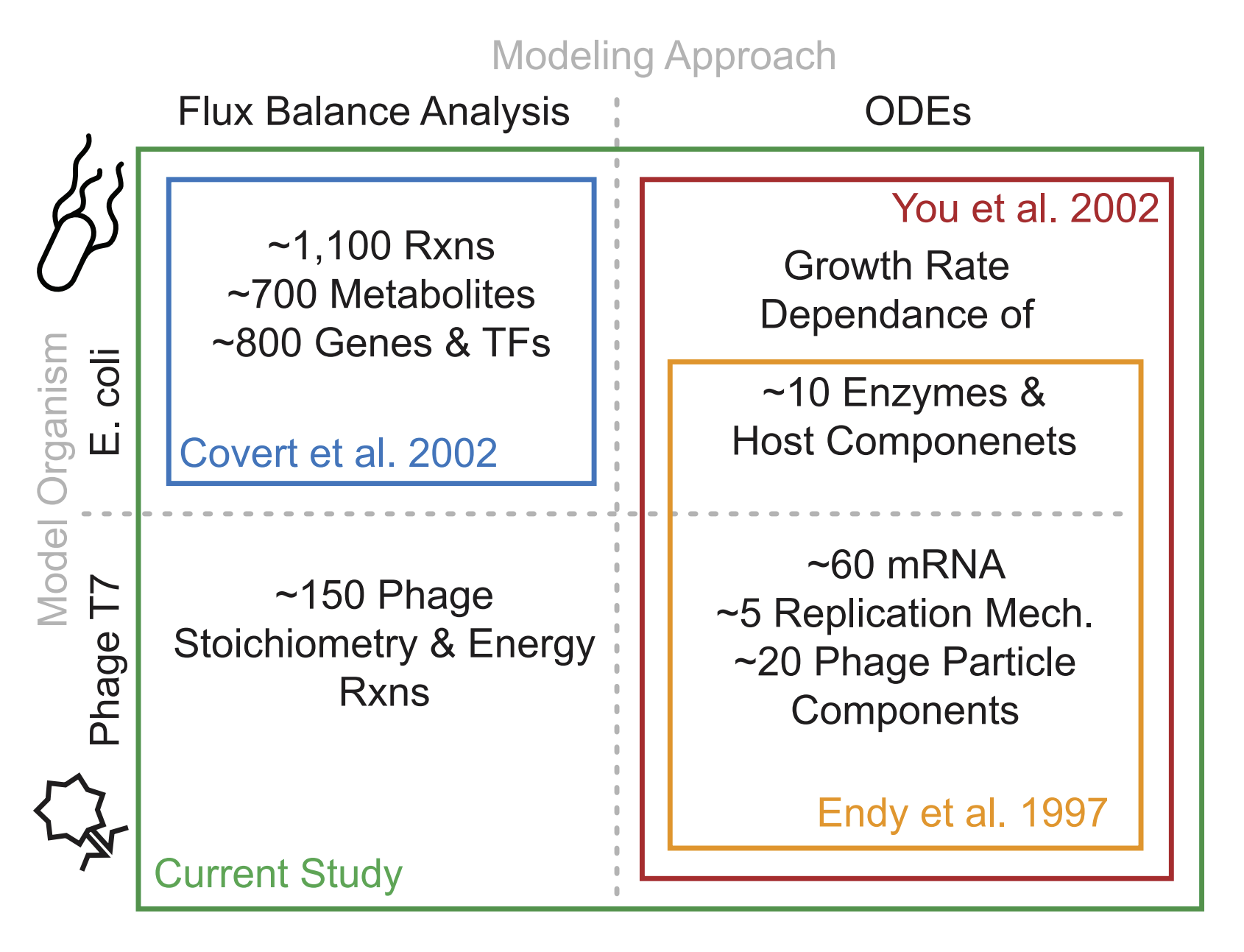
Bacteriophages are optimal candidates to be modeled due to their small genome size and quick life cycles/reproduction rates. The genome of T7 was first sequenced in 1983. Since then, the discovery of a lot of specific features of T7 such as functions of specific genes, RNAse activity, and the location of specific promoters and terminators, has led to many detailed mechanistic models of the T7 viral cycle in a host bacterial cell.
First in 2000, a computer simulation was created to predict and test the organizational efficiency of the T7 genome. This simulation itself was based on experimentally-found data from T7, and was used for the wild type as well as over 100,000 mutant strains. In the mutants, the first gene (gene 1) that codes for T7 RNA polymerase was expressed randomly across the genome, leading to many kinds of different developmental patterns of the T7 phage. The protein expression levels were corroborated with experimental data in order to verify the validity of the model (found to be moderately equivalent to the experimental data). This resulted in the determination that the wild-type T7 genome is almost optimal for quick development, as only 2.8% of the mutants were found to grow more quickly (Endy et al. 1997, 2000).
Next in 2007, a new model called TABASCO was used to compute genome-wide gene expression while allowing for the “representation of individual molecules and events in gene expression”. This fine-scale simulator was used to simulate the entirety of T7 gene expression throughout the infection cycle. This study was able to confirm that the "single molecule, base-pair resolved" model using TABASCO was accurately able to simulate gene expression, and that this model was able to distinguish itself from previous models by having the capacity to directly account for intermolecular events such as "polymerase collisions" and "promoter occulusion" due to its fine-scale modeling (Kosuri, Kelly, and Endy 2007).
In 2012, two separate models: a set of “structured ordinary differential equations” (ODEs) that quantified T7 replication and “E. coli flux balance analysis metabolic model” (FBA) were integrated in order to model the T7 replication in E. coli. This integrated model enabled the prediction of progeny virion production based on only the specification of host cell nutritional environment, and the results were found to be “favorable” when compared to experimentally-derived data. Overall, this model shows that the metabolic limitation of the host is just as important in T7 replication dynamics as the number of proteins synthesized by the phage, which is was a novel breakthrough at the time. This integrated model was also able to explain virus infection dynamics in different cultural mediums in a consistent way that neither of the previous models could accomplish separately (Birch, Ruggero, and Covert 2012).

Pinetree and the Gillespie Algorithm
One of the drawbacks of using differential equations to model biochemical systems is that they assume a large concentration of reagents so that millions of reactions are occurring simultaneously. In real biology, the reagents in stochastic processes are often much less concentrated, causing these models to overestimate product creation. In order to combat this, Pinetree employs the Gillespie Stochastic Model Algorithm, a mathematical model that simulates the effects of individual molecules in stochastic mechanisms. By tracking interactions at the molecular level, the Gillespie algorithm provides a much more accurate picture of the processes that are going on inside of individual cells.
The Gillespie Algorithm follows the following structure:
- Initialize. The simulation specifies the names and quantities of all free species and sets the time point to zero. Additionally, it creates a set of reactions that these species can participate in, assigning each with its respective rate constant.
- Determine Propensities. The simulation assigns a propensity to each reaction. This number determines how likely that reaction is to occur at the current time step, and is determined by the number of free species available to engage in said reaction.
- Generate Randomness. Also know as the Monte Carlo step (after another algorithm that Gillespie draws heavily from), the simulation randomly chooses a reaction to occur (weighted on propensity) and calculates a random time step to wait before the next reaction occurs. In Pinetree, this time step is calculated using the sum of all reaction propensities.
- Update. The simulation executes the chosen reaction and advances the time point by the amount generated in the previous step.
- Iterate. Steps 2-5 are repeated until the time point surpasses a preset value.
This algorithm is capable of accurately modeling huge numbers of interactions at the molecular level with a relatively small amount of processing power, and thus is an excellent tool for investigating the biochemical processes that occur during a phage infection.
Computational Implementation
Pinetree separates its simulation of individual chemical species from their reactions using a distinct SpeciesTracker class and a family of Reaction classes, respectively. The SpeciesTracker loads in each species specified in the model and tracks its copy number throughout the simulation. SpeciesTracker also maintains species-to-Reaction maps that allow the Gillespie model to cache reaction propensities until new ones are required to be calculated, saving on processing power. Polymers are managed as a subclass of Reactions that contain objects corresponding to bound polymerases, as well as genomic features such as promoters, terminators, and RNAse sites. The polymerase objects track their position within the polymer they're bound to. The binding event itself is managed by another reaction subclass, Bind, that removes polymerases from the SpeciesTracker pool and adds them to polymers.

Modeling T7 Bacteriophage
Pinetree was originally used by Benjamin Jack to examine the effects of mRNA transcript degradation and codon usage on the expression of the T7 bacteriophage genome. We further utilized this model to examine the changes in expression that resulted from various mutations we applied to the genome. These mutations included sequential deletions of genes, addition of sfGFP in various locations in the genome, changing the position and copy number of the gene responsible for holin protein creation, and the deletion of RNAse degradation sites. We also modified the T7 model in order to simulate the effect of a mutation that disabled the T7 lysozyme protein's ability to cooperatively bind T7 polymerase.
Original Pinetree Modelling (Jack et al., 2019)
Most stochastic gene expression simulators rely on an assumption of steady-state mRNA transcript abundances. However, transcripts in real biological systems fluctuate based on the transcription rates of polymerases and the rates at which RNAse enzymes degrade them. Pinetree attempts to simulate this dynamism by incorporating RNAse sites as objects in the polymer class (similar to how promoters are implemented) that could be recognized by RNAse objects, as well as setting a rate constant at which bound RNA is degraded. This change makes Pinetree more accurately model real transcript levels (Jack et al., 2019).

Modified T7 Genomes
When we first started modelling T7's genome with Pinetree, we sought to verify that it was capable of effectively simulating the various types of mutations we wanted to implement onto the T7 genome, including deletions, insertions, and gene movements. To do this, we first recreated a gene expression profile for the wild-type T7 genome. This provided us with a baseline that we could compare the results from our mutated genomes to.

After obtaining a wild-type profile, we wanted to test the simulation's ability to recognize and accurately model mutations dealing with deleted and inserted genes. We found that Pinetree could effectively account for deleted genes with gaps in its expression profile, as well show the expression of newly inserted ones.


After confirming the Pinetree could simulate insertions and deletions of genes with the phage T7 genome, we wanted to find out if it could accurately model the effects of moving genes as well. We know that the T7 genome is unidirectional and utilizes polycistronic RNA transcripts, meaning that the location of genes within the genome (especially in relation to promoters and terminators) has a substantial effect on their expression. To test if Pinetree could account for this, we created a mutant genome wherein gene 17.5 (which codes for a holin protein) was moved to a point directly upstream of gene 10A (which codes for the major capsid protein). This site has several promoters upstream of it, and our previous modeling has indicated that it is the point of highest expression within the genome. We found that after moving gene 17.5 to this highly expressed region, Pinetree showed that it was expressed at similar levels to gene 10A. This validated our hypothesis that Pinetree can effectively model the effects of the genomic organization of promoters and terminators on the expression of various loci.
Calculating Lysis Time and Burst Size
To create the burst size calculator, we first determined how many of each virion protein is present in each T7 virion (Molineux, 2006). Then we created a spreadsheet listing out these numbers which is then fed into the calculator along with the Pinetree output files. The calculator then calculates the number of virions that would be produced based on each virion protein individually. It then takes the minimum of these numbers as our burst size so that it is based on the limiting virion protein.


To create the lysis time calculator, we first found the number of holins present when the WT model should lyse at 1320 seconds in our simulation. This time point was derived from a conversion from "experimental time" to "simuation time. This conversion is required because the rate constants used in our T7 simulation were taken from experiments done at 25 or 30 degrees C, while the experimental data collected in Jack et al. 2019 was done at 37 degrees C. Because T7 bacteriophage lyses at around 11 minutes at 37 degrees C (Jack et al., 2017), a conversion factor was applied to roughly equate 500 seconds in simulation to 5 minutes in vitro, and 1000 seconds in simulation to 9 minutes in vitro. Althought it is not a completely linear correlation, we estimated that the lysis time of 11 minutes became roughly 1320 seconds in simulation time.
Using our wild-type T7 data, we found the average number of holins that existed at 1320 seconds (the estimated lysis time in simulation) to be 12341. We chose to use holins as a benchmark for lysis as their accumulation is responsible for making the cellular membrane more porous, ultimately leading to lysis. Our lysis time calculator takes in expression data from Pinetree and finds the time point in each simulation when holin abundance passes this threshold, and marks that as the lysis time for that simulation. We can then aggregate this data to display the total protein or transcript abundances for each simulation at their individual lysis times, as well as examine how the distribution of lysis time varies between the wild-type and our mutant genomes.


Partnership with the TU Delft iGEM team
One of the important contributors to our project was the TU Delft iGEM team. Through our partnership, we were able to support and improve each other’s projects by considering different modeling perspectives for T7 bacteriophage: population dynamics and gene products. We were able to better understand how to maximize signal production and how we can alter our product to make it more effective in the future.
Overall, the partnership with the TU Delft iGEM team was very successful. Both of our teams gained new perspectives on modeling the T7 bacteriophage and helped each other brainstorm different ideas. From this partnership, we were able to understand that the concentrations of bacterial cells we aim to detect is unlikely to have many cells lyse. Therefore, we need a filter system to increase the concentration of the bacteria to lyse more cells. For the TU Delft iGEM team, we were able to provide insight into their wet lab results and offer recommendations about different locations to place their reporter gene in the T7 bacteriophage genome. We are looking forward to using these suggestions and information that we have learned in future advancements for this project. You can learn more about our work with TU Delft on the Partnership page.
Improving Pinetree
Pinetree is still a relatively new software, and still has many opportunities for improvement. Throughout our project we have had to make several tweaks to the code in order to simulate our mutated T7 genomes, as well as to make it more responsive and useful for achieving our project goals.
Polymerase Collision Error
One of the first major bugs we encountered had to do with the way Pinetree handled polymerase objects and random seeding of variables during initialization. Pinetree usually uses a random seed that helps it determine how to assign random values (such as polymerase locations and reaction propensities) at the beginning of the simulation, however, it also allows for specific seeds to be set to aid in data reproducibility. When we utilized this feature, we found that we were getting an error having to do with a collision of polymerase objects in nearly all of the random seeds we tried. We reported this bug back to the Wilke lab, and they determined that it had something to do with polymerases being created at the same space at the start of simulations and quickly resolved the issue. After this was solved we were able to start collecting data.
RNAse Degradation
One of the more interesting functionalities of Pinetree is the ability to simulate the effect of RNA transcript degradation within a cell. This allows for the simulation to more accurately predict transcript (and thus protein) abundances at later time points, which was especially useful for us when investigating the proteins available at lysis time during a T7 infection. However, after running a few simulations, we found that this function was not turned on by default. After enabling transcript degradation we found that the transcript abundances were much lower than those reported in the original Jack paper. After doing some digging in the methods section, we found that they had to scale up their polymerase speed by a factor of ten in order to account for reduced production that resulted from transcripts being degraded. After making this tweak, our data resembled expected values much more closely. To confirm this activity, we tested genomes that had certain RNAse sites(3.8 and 6.5) removed and saw that genes directly downstream of these sites had much higher expression. While we did not utilize them for our final T7 mutant, we showed that deleting or modifying the strengths of RNAse sites can fine tune transcript abundances for specific regions in the genome.


Lysozyme Mutant
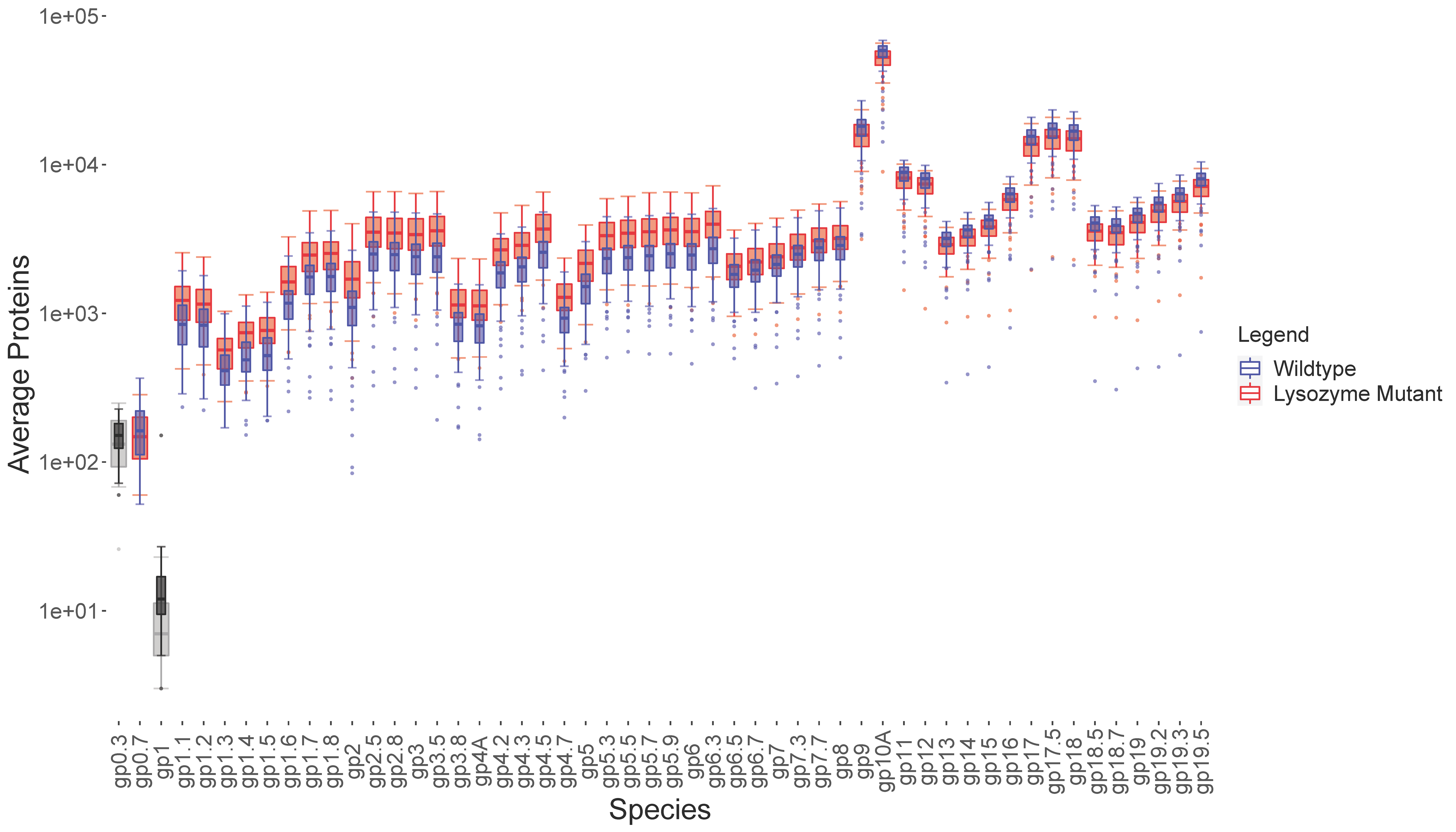
We also investigated the effects of T7 bacteriophage's lysozyme protein on gene expression. In addition to acting as an amidase that degrades the bacterial cell wall, the T7 lysozyme (encoded by gene 3.5) binds to T7 RNA polymerase and inhibits transcription, limiting the amount of class II and III gene transcripts that are created in later stages of an infection. Several lysozyme mutants have been characterized that do not exhibit this regulatory effect (Cheng et al., 1994). We hypothesized that by deleting the interaction between the lysozyme and polymerase objects in our model we could simulate the effect of the mutant lysozyme, resulting in the upregulation of class II and III genes and increasing the amount of GFP created. After running simulations using our mutant lysozyme model, we found that class II genes were indeed upregulated. However, expression of class III genes in the mutant lysozyme model was significantly lower than those in the wild-type. We hypothesized that this may be due to the fact that the upregulation of class II genes takes up more of the RNA polymerases' time and transcript space, meaning there are less available to express class III genes.
Conclusion
Overall, our team effectively used modeling to gain insight on our final design of the ideal mutant T7 bacteriophage genome, which will serve as an efficient, portable reporter for water contamination. Our use of the Pinetree software and our creation of the burst size and lysis time calculators empowered the team to determine the maximum number of virions produced at the optimal lysis time while also maximizing GFP expression for our final T7 bacteriophage genome. Our experimentation with the Pinetree simulation was also able to yield important improvements to the modeling software, as the issues relating to the polymerase collision and RNAse degradation were reported and ultimately fixed to yield more realistic modeling results. Through our modeling and partnerships with other teams, our total modeling data and improvements to the software will enable future researchers to be able to model T7 bacteriophage variations and even other similar phages much more accurately than before. Furthermore, this wealth of data that our team generated will allow the team to construct and test our final T7 phage genome in a wetlab setting during 2021.
References
Birch, E. W., Ruggero, N. A., and Covert, M. W. (2012)‘Determining Host Metabolic Limitations on Viral Replicationvia Integrated Modeling and Experimental Perturbation’.PLoS Computational Biology, 8: e1002746.
Cheng X., Zhang X., Pflugrath J.W., Studier F.W. The structure of bacteriophage T7 lysozyme, a zinc amidase and an inhibitor of T7 RNA polymerase. Proc. Natl. Acad. Sci. USA. 1994;91:4034–4038. doi: 10.1073/pnas.91.9.4034.
Endy et al. (2000) ‘Computation, Prediction, and Experimental Tests of Fitness for Bacteriophage T7 Mutants with Permuted Genomes’, Proceedings of the National Academy of Sciences of the United States of America, 97: 5375–80.
Jack, B. R., and Wilke, C. O. (2019) ‘Pinetree: A Step-Wise GeneExpression Simulator with Codon-Specific Translation Rates’,Bioinformatics, 35: 4176–8.
--- et al. (2017) ‘Reduced Protein Expression in a Virus Attenuated by Codon Deoptimization’, G3 (Bethesda),7:2957–68.
Jack, Benjamin R., Boutz, Daniel R., Paff, Matthew L., Smith, Bartram L., Wilke, Claus O. “Transcript Degradation and Codon Usage Regulate Gene Expression in a Lytic Phage.” Virus Evolution, vol. 5, no. 2, Oxford University Press (OUP), July 2019, pp. vez055–vez055, doi:10.1093/ve/vez055.
Kosuri, S., Kelly, J. R., and Endy, D. (2007) ‘TABASCO: A Single Molecule, Base-Pair Resolved Gene Expression Simulator’, BMCBioinformatics, 8: 480.
Molineux, Ian. "The Bacteriophages." Science, vol. 242, no. 4883, 1988, p. 1317.