Team:UCopenhagen/Results

Introduction
The modular nature of the biosensor enables multiple setups for different purposes. From the implementation point of view, it also creates multiple points for optimization and verification. In order to verify that all of our different modules work we divided designs into smaller constituents and created experiments around them in order to test their functionality. This was done in parallel with creating and assaying our complete whole cell biosensors.
To lay out what experiments we had to do, we first had to think of the pitfalls of each of the modules, and what could go wrong in different scenarios. Therefore, we chose the following questions to guide our choice of experiments:
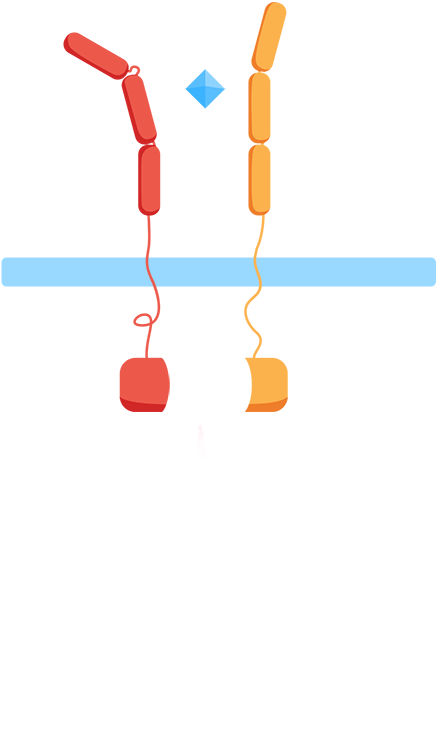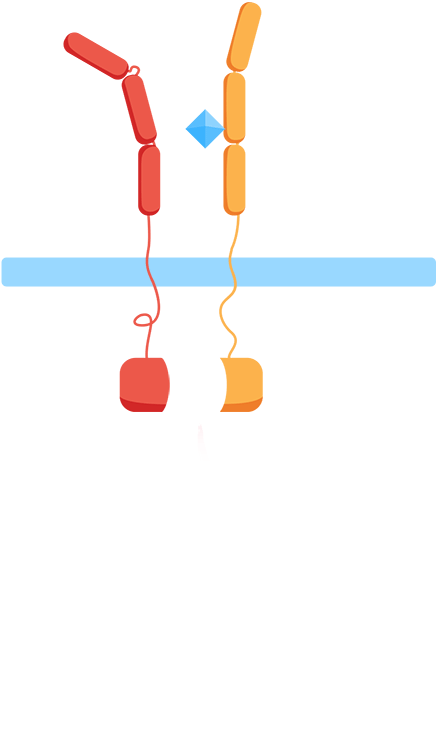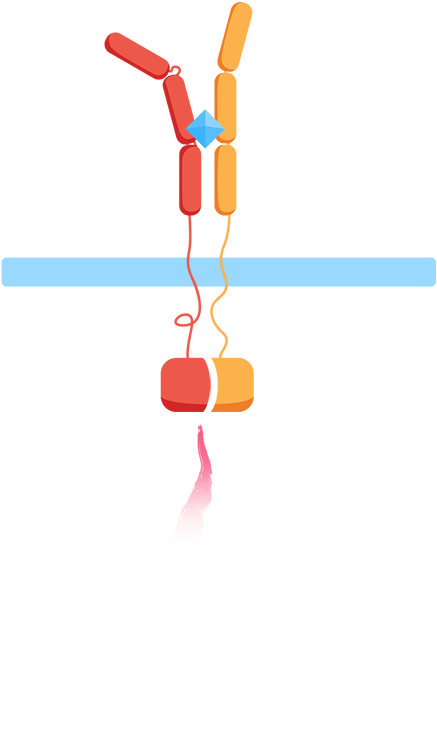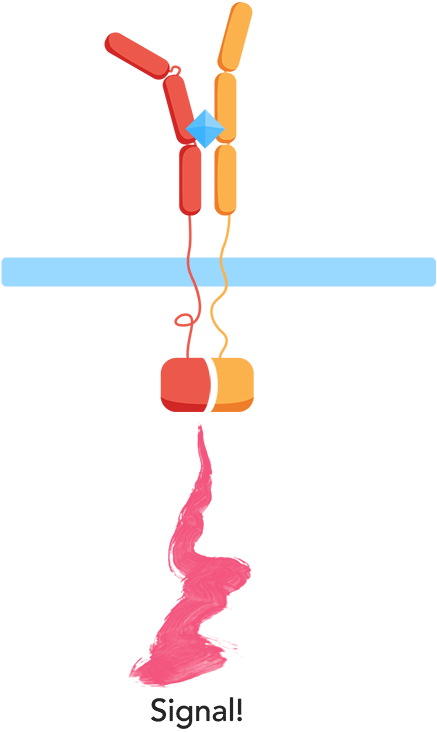
- ✧ Are the extracellular domains functioning as intended? Are they dissociated in the absence of the interleukins and will they associate in the presence of the interleukins?
- ✧ Do the receptors and accessory proteins have the intended subcellular localization?
- ✧ Is the membrane bound transcription factor of the intermediate design able to be cleaved by a TEV protease?
- ✧ Is the TEV protease able to cleave the modified Gα (Gpa1p) proteins and trigger the yeast pheromone pathway?
- ✧ Do the modified Gα (Gpa1p) proteins retain their ability to transduce signalling from a GPCR?
To answer these questions we followed the scientific workflow shown in figure 2.

Cloning

Then, the fragments were digested with the USER enzyme to create custom sticky ends. These fragments were ligated by incubation with the relevant linearized vector. Subsequently, competent E. coli cells were transformed with the products of the USER ligation and grown on agar plates with carbenicillin. This allowed for selection for cells that had been transformed with plasmids from our USER ligation since all our plasmid backbones contained a gene encoding β-lactamase (AmpR).



|
Construct name |
Vector |
Promoter |
Domains in gene |
Status |
|
Sgp130(D1-D6)-Cub |
X3A |
pCCW12 |
SP-sgp130(D1-D6)-TMD-Cub-LexA-VP16 |
Done |
|
Sgp130(D1-D6)-Cub |
X3A |
pCCW12 |
SP-sgp130(D1-D6)-TMD-Cub-LexA-VP16-sfGFP |
Done |
|
Sgp130(D1-D6)-Nub |
Ass2C |
pTDH3 |
SP-Sgp130(D1-D6)-TMD-Nub |
Done |
|
Sgp130(D1-D6)-Nub |
Ass2C |
pTDH3 |
SP-Sgp130(D1-D6)-TMD-Nub-sfGFP |
No - two colonies had mutations did not sequence third |
|
sIL-6R (secreted) |
Ass2C |
pPGK1 |
Alpha factor-pre-pro-sequence -sIL-6R |
Done |
|
sIL-6R (secreted) |
Ass2B |
pPGK1 |
Alpha factor-pre-pro-sequence -sIL-6R |
Done |
|
sIL-6R (secreted) |
Ass2C |
pPGK1 |
Alpha factor-pre-pro-sequence -sIL-6R |
Done |
|
Sgp-130(D1-D3)-Cub |
X3A |
pCCW12 |
SP-Sgp130(D1-D3)-TMD-Cub-LexA-VP16 |
Done |
|
Sgp-130(D1-D3)-Cub |
X3A |
pCCW12 |
SP-Sgp130(D1-D3)-TMD-Cub-LexA-VP16-sfGFP |
Done |
|
sIL-6R-Nub |
Ass2C |
pTDH3 |
SP-sIL-6R-TMD-Nub |
Done |
|
sIL-6R-Nub |
Ass2C |
pTDH3 |
SP-sIL-6R-TMD-Nub-sfGFP |
Done |
|
sIL-1R2-Cub |
X3A |
pCCW12 |
SP-sIL-1R2-TMD-Cub-LexA-VP16 |
Done |
|
sIL-1R2-Cub |
X3A |
pCCW12 |
SP-sIL-1R2-TMD-Cub-LexA-VP16-sfGFP |
Done |
|
sIL-1RAcP-Nub |
Ass2C |
pTDH3 |
SP-sIL-1RAcP-TMD-Nub |
Done |
|
sIL-1RAcP-Nub |
Ass2C |
pTDH3 |
SP-sIL-1RAcp-TMD-Nub-sfGFP |
Redo USER ligation |
|
sIL-10R1-Cub |
X3A |
pCCW12 |
SP-sIL-10R1-TMD-Cub-LexA-VP16 |
Done |
|
sIL-10R1-Cub |
X3A |
pCCW12 |
SP-sIL-10R1-TMD-Cub-LexA-VP16-sfGFP |
Done |
|
sIL-10R2-Nub |
Ass2C |
pTDH3 |
SP-sIL-10R2-TMD-Nub |
Done |
|
sIL-10R2-Nub |
Ass2C |
pTDH3 |
SP-sIL-10R2-TMD-Nub-sfGFP |
Done |
|
MJDOD |
X3C |
LexAop |
Already in lab |
Already in lab |
|
Sgp-130(D1-D6)-CTEV |
X3A |
pCCW12 |
SP-Sgp-130(D1-D6)-TMD-CTEV |
Done |
|
Sgp-130(D1-D6)-NTEV |
Ass2C |
pTDH3 |
SP-Sgp-130(D1-D6)-TMD-NTEV |
Done |
|
Sgp-130(D1-D6)-CTEV |
X3A |
pCCW12 |
SP- Sgp-130(D1-D6)-TMD-CTEV-sfGFP |
Done |
|
Sgp-130(D1-D6)-NTEV |
Ass2C |
pTDH3 |
SP- Sgp-130(D1-D6)-TMD-NTEV-sfGFP |
Done |
|
Sgp130-(D1-D3)-CTEV |
X3A |
pCCW12 |
SP-Sgp130-(D1-D3)-TMD-CTEV |
Done |
|
sIL-6R-NTEV |
Ass2C |
pTDH3 |
SP-sIL-6R-TMD-NTEV |
Done |
|
Sgp130(D1-D3)-CTEV |
X3A |
pCCW12 |
SP-sgp130-TMD-CTEV-sfGFP |
USER-ligation produced stop codon.
|
|
sIL-6R-NTEV |
Ass2C |
pTDH3 |
SP-sIL-6R-TMD-NTEV-sfGFP |
Done |
|
sIL-1R2-CTEV |
X3A |
pCCW12 |
SP-sIL-1R2-TMD-CTEV |
Done |
|
sIL-1RAcp-NTEV |
Ass2C |
pTDH3 |
SP-sIL-1RAcp-TMD-NTEV |
Done |
|
sIL-1R2-CTEV |
X3A |
pCCW12 |
SP-sIL-1R2-TMD-CTEV-sfGFP |
USER-ligation produced stop codon. New primer needed |
|
sIL-1RAcP-NTEV |
Ass2C |
pTDH3 |
SP-sIL-1RAcP-TMD-NTEV-sfGFP |
Done |
|
sIL-10R1-CTEV |
X3A |
pCCW12 |
SP-sIL-10R1-TMD-CTEV |
Done |
|
sIL-10R2-NTEV |
Ass2C |
pTDH3 |
SP-sIL-10R2-TMD-NTEV |
Done |
|
sIL-10R1-CTEV |
X3A |
pCCW12 |
SP-sIL-10R1-TMD-CTEV-sfGFP |
USER-ligation produced stop codon.
|
|
sIL-10R2-NTEV |
Ass2C |
pTDH3 |
SP-sIL-10R2-TMD-NTEV-sfGFP |
Done |
|
E3-CTEV (BBa_K1159102) |
X3A |
SP-E3-TMD-CTEV(BBa_K1159102) |
Do colony PCR |
|
|
ZE-CTEV (BBa_K1159102) |
SP-ZE-TMD-CTEV (BBa_K1159102) |
Do colony PCR |
||
|
K3-NTEV |
SP-K3-TMD-NTEV |
Do colony PCR |
||
|
ZR-NTEV |
SP-ZR-TMD-NTEV |
Do colony PCR |
||
|
E3-CTEV (truncated C-terminal) |
SP-E3-TMD-CTEV(truncated C-terminal) |
Do colony PCR |
||
|
ZE-CTEV (truncated C-terminal) |
SP-ZE-TMD-CTEV (truncated C-terminal) |
Do colony PCR |
||
|
E3-CTEV (wt) |
SP-E3-TMD-CTEV(wt) |
Do colony PCR |
||
|
ZE-CTEV (wt) |
SP-ZE-TMD-CTEV (wt) |
Do colony PCR |
||
|
40 |
cancelled |
|||
|
41 |
cancelled |
|||
|
42 |
cancelled |
|||
|
43 |
cancelled |
|||
|
44 |
Ass2A |
pPGK1 |
SP-TMD-ENLYFQG-LexA-VP16 |
Done |
|
45 |
cancelled |
|||
|
46 |
Ass2a |
ppgk1 |
SP-TMD-ENLYFQG-LexA-VP16-sfGFP |
Done |
|
47 |
cancelled |
|||
|
48 |
new plan for G-alpha |
|||
|
49 |
new plan for G-alpha |
|||
|
50 |
new plan for G-alpha |
|||
|
51 |
Ass2A |
pPGK1 |
already in lab |
|
|
52 |
Ass2C |
scGAL1 |
TEV protease |
done |
|
53 |
Ass2B |
pRET |
Ste12 |
cancelled |
|
54 |
Ass2B |
pRET |
LexA-Ste12 |
already in lab |
|
55 |
Ass2B |
pFIG |
LexA-nanoluciferase |
already in lab |
|
56 |
X3A |
pCCW12 |
A2A (R199A) |
already in lab |
|
GPA1 mutations: |
||||
|
GPA1mut1 |
Ass2A |
pPGK1 |
GPA1 with cut site inserted at site one |
done |
|
GPA1mut2 |
Ass2A |
pPGK1 |
GPA1 with cut site inserted at site two |
done |
|
GPA1mut3 |
Ass2A |
pPGK1 |
GPA1 with cut site inserted at site three |
done |
|
GPA1mut4 |
Ass2A |
pPGK1 |
GPA1 with cut site inserted at site four |
done |
|
GPA1mut5 |
Ass2A |
pPGK1 |
GPA1 with cut site inserted at site five |
done |
Yeast strains



|
Assembly nr. |
Description |
X3A |
Ass2A |
Ass2B |
Ass2C |
X3C |
Status |
|
CD1 |
IL-10 biosensor using IL-10RI and IL-10II and the split ubiquitin |
Il-10RI-Cub (15) |
empty |
empty |
Il-10RII-Nub (17) |
LexAop-nanoluc |
Confirmed |
|
CD2 |
IL-6 biosensor using the IL-6R and truncated sgp130 and split ubiquitin design |
sgp130(D1-D3)-Cub (7) |
empty |
empty |
sIL-6R-Nub (9) |
LexAop-nanoluc |
Confirmed |
|
CD3 |
IL-1 biosensor using IL-1R2 and IL-1RAcP and the split ubiquitin design |
sIL-1R2-Cub (11) |
empty |
empty |
sIL-1RAcP-Nub (13) |
LexAop-nanoluc |
Confirmed |
|
CD4 |
IL-6 biosensor using the IL-6R and truncated sgp130 and the split-TEV and TMD-LexA-VP16 design |
sgp130(D1-D3)-CTEV (24) |
TMD-LexA-VP16 (44) |
empty |
sIL-6R-NTEV (25) |
LexAop-nanoluc |
no |
|
CD5 |
IL-1 biosensor using IL-1R2 and IL-1RAcP and the split-TEV and TMD-LexA-VP16 design |
sIL-1R2-CTEV (28) |
TMD-LexA-VP16 (44) |
empty |
sIL-1RAcP-NTEV (29) |
LexAop-nanoluc |
Confirmed |
|
CD6 |
IL-10 biosensor using IL-10RI and IL-10II and the split-TEV and TMD-LexA-VP16 design |
sIL-10RI-CTEV (32) |
TMD-LexA-VP16 (44) |
empty |
sIL-10RII-NTEV (33) |
LexAop-nanoluc |
no |
|
CD7 |
IL-6 biosensor using the IL-6R and truncated sgp130 and the split-TEV protease and Gα cleavage design |
sgp130(D1-D3)-CTEV (24) |
GPA1-mut2 |
LexA-Ste12 |
sIL-6R-NTEV (25) |
LexAop-nanoluc |
no |
|
CD8 |
IL-1 biosensor using IL-1R2 and IL-1RAcP and the split-TEV protease and Gα cleavage design |
sIL-1R2-CTEV (28) |
GPA1-mut2 |
LexA-Ste12 |
sIL-1RAcP-NTEV (29) |
LexAop-nanoluc |
no |
|
CD9 |
IL-10 biosensor using IL-10RI and IL-10II and the split-TEV protease and Gα cleavage design |
sIL-10RI-CTEV (32) |
GPA1-mut2 |
LexA-Ste12 |
sIL-10RII-NTEV (33) |
LexAop-nanoluc |
no |
|
CD16 |
IL-6 biosensor using sgp130 and secreted sIL-6R and split ubiquitin design |
sgp130(D1-D6)-cub (1) |
empty |
sIL-6R (5) (ass2B) |
sgp130(D1-D6)-Nub (3) |
LexAop-nanoluc |
Confirmed |
|
CD17 |
IL-6 biosensor using sgp130 and secreted sIL-6R and the split-TEV and TMD-LexA-VP16 design |
sgp130(D1-D6)-cTEV (20) |
TMD-TF (44) |
sIL-6R (5) (ass2B) |
sgp130(D1-D6)-nTEV (21) |
LexAop-nanoluc |
no |
|
CD18 |
IL-6 biosensor using sgp130 and secreted sIL-6R and the split-TEV protease and Gα cleavage design |
sgp130(D1-D6)-cTEV (20) and sgp130(D1-D6)-nTEV (21) (57) |
GPA1 mut |
LexA-Ste12 |
sIL-6R (5) |
LexAop-nanoluc |
no |
|
Assembly nr. |
Description |
X3A |
Ass2A |
Ass2B |
Ass2C |
X3C |
Status |
|
CD19 |
For succellular localization assay of sgp130(D1-D6)-Cub |
sgp130(D1-D6)-Cub-sfGFP (2) |
empty |
empty |
empty |
empty |
Confirmed |
|
CD20 |
For succellular localization assay of sgp130(D1-D6)-Nub |
empty |
empty |
empty |
sgp130(D1-D6)-Nub-sfGFP (4) |
empty |
no |
|
CD21 |
For succellular localization assay of sgp130(D1-D3)-Cub |
sgp130(D1-D3)-Cub-sfGFP (8) |
empty |
empty |
empty |
empty |
Confirmed |
|
CD22 |
For succellular localization assay of sIL-6R-Nub |
empty |
empty |
empty |
sIL-6R-Nub-sfGFP (10) |
empty |
Confirmed |
|
CD23 |
For succellular localization assay of sIL-1R2-Cub |
sIL-1R2-Cub-sfGFP (12) |
empty |
empty |
empty |
empty |
Confirmed |
|
CD24 |
For succellular localization assay of sIL-1RAcP-Nub |
empty |
empty |
empty |
sIL-1RAcP-Nub-sfGFP (14) |
empty |
no |
|
CD25 |
For succellular localization assay of sIL-10R1-Cub |
sIL-10R1-Cub-sfGFP (16) |
empty |
empty |
empty |
empty |
Confirmed |
|
CD26 |
For succellular localization assay of sIL-10R2-Nub |
empty |
empty |
empty |
sIL-10R2-Nub-sfGFP (18) |
empty |
Confirmed |
|
CD27 |
For succellular localization assay of sgp130(D1-D6)-cTEV |
sgp130(D1-D6)-cTEV-sfGFP (22) |
empty |
empty |
empty |
empty |
Confirmed |
|
CD28 |
For succellular localization assay of sgp130(D1-D6)-nTEV |
empty |
empty |
empty |
sgp130(D1-D6)-nTEV-sfGFP (23) |
empty |
Confirmed |
|
CD29 |
For succellular localization assay of sgp130(D1-D3)-cTEV |
sgp130(D1-D3)-cTEV-sfGFP (26) |
empty |
empty |
empty |
empty |
no |
|
CD30 |
For succellular localizationassay of sIL-6R-nTEV |
empty |
empty |
empty |
sIL-6R-nTEV-sfGFP (27) |
empty |
Confirmed |
|
CD31 |
For succellular localization assay of sIL-1R2-cTEV |
sIL-1R2-cTEV-sfGFP (30) |
empty |
empty |
empty |
empty |
no |
|
CD32 |
For succellular localization assay of sIL-1RAcP-nTEV |
empty |
empty |
empty |
sIL-1RAcP-nTEV-sfGFP (31) |
empty |
Confirmed |
|
CD33 |
For succellular localization assay of sIL-10R1-cTEV |
sIL-10R1-cTEV-sfGFP (34) |
empty |
empty |
empty |
empty |
no |
|
CD34 |
For succellular localization assay of sIL-10R2-nTEV |
empty |
empty |
empty |
sIL-10R2-nTEV-sfGFP (35) |
empty |
Confirmed |
|
CD35 |
For succellular localization assay of sIL-6R |
empty |
empty |
empty |
sIL-6R-sfGFP (6) |
empty |
no |
|
CD36 |
For succellular localization assay of TMD-LexA-VP16 |
empty |
TMD-TF-sfGFP (46) |
empty |
empty |
empty |
no |
|
Assembly nr. |
Description |
X3A |
Ass2A |
Ass2B |
Ass2C |
X3C |
Status |
|
CD12.01 |
A2A biosensor with GPA1-mutation 1 |
A2A (R199A) |
GPA1-mutation 1 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD12.02 |
A2A biosensor with GPA1-mutation 2 |
A2A (R199A) |
GPA1-mutation 2 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD12.03 |
A2A biosensor with GPA1-mutation 3 |
A2A (R199A) |
GPA1-mutation 3 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD12.04 |
A2A biosensor with GPA1-mutation 4 |
A2A (R199A) |
GPA1-mutation 4 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD12.05 |
A2A biosensor with GPA1-mutation 5 |
A2A (R199A) |
GPA1-mutation 5 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD13 |
A2A biosensor |
A2A (R199A) |
GPA1 |
LexA-Ste12 |
empty |
LexAop-nanoluc |
no |
|
CD14.01 |
A2A biosensor with GPA1-mutation 1 and inducible TEV protease |
A2A (R199A) |
GPA1-mutation1 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
Confirmed |
|
CD14.02 |
A2A biosensor with GPA1-mutation 2 and inducible TEV protease |
A2A (R199A) |
GPA1-mutation2 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
Confirmed |
|
CD14.03 |
A2A biosensor with GPA1-mutation 3 and inducible TEV protease |
A2A (R199A) |
GPA1-mutation3 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
Confirmed |
|
CD14.04 |
A2A biosensor with GPA1-mutation 4 and inducible TEV protease |
A2A (R199A) |
GPA1-mutation4 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
no |
|
CD14.05 |
A2A biosensor with GPA1-mutation 5 and inducible TEV protease |
A2A (R199A) |
GPA1-mutation5 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
no |
|
CD15 |
A2A biosensor and inducible TEV protease |
A2A (R199A) |
GPA1 |
LexA-Ste12 |
scGal1-TEV |
LexAop-nanoluc |
Confirmed |
|
Assembly nr. |
Description |
X3A |
Ass2A |
Ass2B |
Ass2C |
X3C |
Status |
|
CD10 |
TMD-LexA-VP16 cleaved by inducible TEV |
empty |
TMD-LexA-VP16 (44) |
empty |
scGal1-TEV |
LexAop-nanoluc |
Confirmed |
|
CD37 |
Leucinezippers (K3 and E3) with partially optimised cTEV as in Bba_K1159102 |
(36.1) |
TMD-LexA-VP16 (44) |
empty |
(37.1) |
LexAop-nanoluc |
no |
|
CD38 |
Leucinezippers (K3 and E3) with most optiimsed cTEV |
(38.1) |
TMD-LexA-VP16 (44) |
empty |
(37.1) |
LexAop-nanoluc |
no |
|
CD39 |
Leucinezippers (K3 and E3) with Wt cTEV as in Bba_K1965020 |
(39.1) |
TMD-LexA-VP16 (44) |
empty |
(37.1) |
LexAop-nanoluc |
no |
|
CD40 |
Leucinezippers (ZR and ZE) with partially optimised cTEV as in Bba_K1159102 |
(36.2) |
TMD-LexA-VP16 (44) |
empty |
(37.2) |
LexAop-nanoluc |
no |
|
CD41 |
Leucinezippers (ZR and ZE) with optimised cTEV |
(38.2) |
TMD-LexA-VP16 (44) |
empty |
(37.2) |
LexAop-nanoluc |
no |
|
CD42 |
Leucinezippers (ZR and ZE) with Wt cTEV as in Bba_K1965020 |
(39.2) |
TMD-LexA-VP16 (44) |
empty |
(37.2) |
LexAop-nanoluc |
no |
Other strains:
|
Assembly nr. |
Description |
X3A |
Ass2A |
Ass2B |
Ass2C |
X3C |
Status |
|
CD11 |
control for reporter leakage |
empty |
empty |
empty |
empty |
LexAop-nanoluc |
no |

Minimal biosensor design
In the minimal design, the extracellular Interleukin-receptors are fused with either a C- or N-terminal part of a split-ubiquitin protein. Upon receptor association in the presence of IL-molecules, the two halves reconstitute resulting in full-length ubiquitin that can be recognized by a deubiquitinase leading to cleavage of the protein and release of a LexA-VP16 synthetic transcription factor. This would further result in the expression of the luciferase reporter gene (nanoluc).

Induction assays
Biosensors for IL-10 and IL-6, corresponding to assembly 1 and 2 in table 2, were tested. The two strains were grown to an OD600=0,5 and incubated at 30°C for a set of different incubation times with different concentrations of the respective biomarker. Next, luciferase assays were performed with the strains (figures 9 and 10).


Interestingly, we noticed that the level of luminescence obtained for the IL-6 biosensor is 3-10 times higher than for the IL-10 biosensor. This suggests that the IL-6 biosensor might be constitutively signalling. One possible explanation is that there was affinity between the two receptor proteins even when the interleukin was not present. This would further infer that the two receptor proteins were localized to the same subcellular compartments. Alternatively, the receptor protein with the transcription factor had been partially degraded which would allow the transcription factor to activate expression of the reporter.
For both biosensors, the amount of luminescence varied over time with the highest intensity recorded after 14 hours of incubation and lower intensity at shorter and longer incubation times. This might reflect that the cells were in different growth phases at the different times of the luciferase assays.
Subcellular localization of receptors
Yeast strains that constitutively express super-folding green fluorescent protein (sfGFP) linked to the C-terminal of the receptor proteins were constructed (see table with strains for localization assays) to track the localization of the proteins. After confirming the strains for chromosomal integration of the receptor-GFP fusion constructs, the strains were grown in liquid media and observed with confocal fluorescence microscopy.
IL-6 receptor proteins
Flourescence Brightfield

However, findings from the confocal fluorescence microscopy cannot lead to rejection of the other hypothesis – that the system is always signalling due to the partial degradation of one of the proteins, thus, allowing the transcription factor to reach the nucleus at all times. Results from the confocal fluorescence microscopy indicate that the problems with the dysfunctional biosensor might be caused by unsatisfactory localization to the plasma membrane.
IL-10 receptor proteins
Flourescence Brightfield

Intermediate biosensor design

Localization of the receptors
Strains constitutively expressing superfolder green fluorescent protein linked to the C-terminal of the receptor proteins were created (table 2), grown and observed with confocal laser scanning microscopy.
Receptor proteins
Flourescence Brightfield


sIL-6R-nTEV seems to localize as desired and could be possibly used as part of a IL-6 biosensor. In contrast, improvement in the localization of sIL-10R2-nTEV may be needed before it can perform its role in a IL-10 biosensor. Likewise, localization of sIL-1RAcP-nTEV needs to be significantly improved before it can function in a biosensor for IL-1.
Cleavage of membrane bound transcription factor
In the intermediate biosensor design, a split TEV protease releases transcription factors from the membrane upon receptor association and TEV reconstitution. For this, we constructed a transmembrane protein - an accessory protein with a transcription factor linked to a transmembrane domain through a flexible linker that had a cut site for the TEV protease. This experiment was designed in order to verify that a TEV protease could induce signalling by cleaving the flexible linker and releasing the transcription factor. For the experiment, we integrated the constitutively expressed accessory protein, an inducible TEV protease and a nanoLuc luciferase under the control of a synthetic LexAop promoter into S. cerevisiae as described above (see assembly 10 in table 2). The strain was grown to an OD600 of 5.1 and incubated at 30°C for 17 hours in either glucose or galactose + raffinose media to repress or activate the transcription of the TEV protease, respectively. Then, a luciferase assay was carried out with the culture.

Advanced biosensor design

Testing functionality of modified GPA1
The different GPA1 mutants were tested in the context of an adenosine biosensor employing the human adenosine GPCR receptor A2A(R199A) (unpublished) in S. cerevisiae, previously developed in the Kampranis lab. Compared to the original biosensor, the strains in this experiment had an inducible TEV protease and the wt GPA1 was exchanged with the modified GPA1 proteins (see table 2, strains 14.1-14.3 and 15). The strains were grown to an OD600 of 0.4 and incubated in either glucose or galactose + raffinose media at 30°C for 17 hours. Next, the amount of signalling was measured with a luciferase assay.

Biosensors with version one and three of the GPA1 subunit showed very high amounts of luminescence compared to the biosensor with wt GPA1 even in glucose media. This might suggest that these versions of GPA1 are always causing signalling through the pheromone pathway. Mutation 2 showed very low amounts of luminescence compared to the biosensor with wt GPA1 even in galactose media. This might suggest that the Gα strongly inhibits all signalling, that the pheromone pathway has been shut down, or that the yeast has mutated so that the reporter expression is uncoupled from the pheromone pathway.
For the biosensor with wt GPA1, a correlation between adenosine concentration and luminescence was observed at a level that reproduced earlier results from the Kampranis lab when glucose was used as a carbon source. A similar tendency was not observed for any of the mutant GPA1 strains neither with glucose nor galactose + raffinose media. This infers that all the mutations cause a loss-of-function in the sense that the GPA1 is not able to transduce the signal from the A2A GPCR.
The comparative experiment of effects of glucose media and galactose + raffinose media on mutant GPA1 strains and the wt GPA1 strain yielded mixed results. It was not possible to confirm nor reject whether TEV protease can trigger signalling via cleavage of mutant GPA1. However, the experiment did show that mutant GPA1 strains lost the ability to transduce signal from the A2A GPCR, while it was maintained in the wildtype GPA1.
Conclusion/discussion
- ✧ Test the functionality of our minimal design biosensor
- ✧ Investigate the subcellular localization of 7 of our receptor proteins
- ✧ Verify the functionality of the accessory protein, the membrane bound synthetic transcription factor, in the intermediate design.
- ✧ Test various functions of the modified GPA1 proteins for the advanced design, eg. the ability to signal after being cleaved by a TEV protease and the ability to transduce the signal from a GPCR.
- ✧ Clone 54 constructs
- ✧ Create 21 new yeast strains
Our induction assays showed that the IL-6 and IL-10 biosensors of our minimal design were not able to sense different concentrations of the respective biomarkers. For the interleukin-6 biosensor, the system seemed to be constantly signalling. This might indicate that the dysfunctionality could, to some extent, be explained by an affinity between the receptor proteins in the absence of IL-6. To understand why our biosensors were dysfunctional, the subcellular localization of our modified receptors and accessory proteins were investigated using confocal fluorescence microscopy. In order for our receptor proteins to work, they had to be localized at the plasma membrane of S. cerevisiae. While none of the modified proteins solely localized to the plasma membrane, they were all expressed to some extent, some more than others. For the minimal design, sgp130(D1-D3)-Cub-sfGFP only localized faintly to the plasma membrane, while the soluble sIL-6R-Nub-sfGFP almost solely was found in inclusion bodies. This might explain the dysfunctionality of the minimal IL-6 biosensor.
sIL-10R1-Cub-sfGFP localized primarily to the plasma membrane and endoplasmic reticulum but sIL-10R2-Nub-sfGFP localized unspecifically to the entire cell, mainly in the cytosolic compartment. This might, therefore, also explain the dysfunctionality of the minimal IL-10 biosensor.
The subcellular localization assays of sIL-6R-nTEV-sfGFP, sIL-1RAcP-nTEV-sfGFP, and sIL-10R2-nTEV-sfGFP all showed cytosolic localization and the presence of inclusion bodies, with only sIL-6R-nTEV-sfGFP localizing primarily to the plasma membrane in cells lacking inclusion bodies. However, this was seen for cells containing vacuoles, which may not be representative for the entire population of cells.
For the intermediate design, it was evident that the transcription factor in the form of LexA-VP16 containing a transmembrane domain and a TEV-recognition site in a flexible linker, was able to be cleaved by a TEV protease. Upon induction of TEV protease expression, and carrying out a luciferase assay, luminescence was increased over 1000-fold. This indicated that the protein functioned as intended. In hindsight, the experiment could have been designed to include a control strain without the inducible TEV protease as well as a strain without the membrane bound transcription factor. A western blot of a tagged version of the protein after incubation in either media could further verify that the protein is cleaved by the TEV protease. The next step in characterizing the membrane bound transcription factor would be to investigate whether it can be cleaved by a membrane bound split TEV protease.
For the advanced design, the functionality of the modified GPA1 proteins was compared to the functionality of the wt GPA1 in an adenosine biosensor. To test how cleavage of the modified GPA1 proteins affected signalling through the pheromone pathway, expression of a TEV protease was controlled by changing media from glucose to galactose. The signalling increased in all the modified GPA1 proteins as expected. Surprisingly, the biosensor with wt GPA1 also showed increased signalling upon change of the media. It is, therefore, inconclusive whether the GPA1 proteins functioned as intended. To shed further light on the functionality of the GPA1 proteins, the experiment would have to be designed differently. This could include making a control strain without the TEV protease or utilizing another method for inducing expression of the TEV protease where the carbon source is kept constant. One possible explanation for the elevated level of luminescence with galactose media is that the pPGK1 promoter, which regulates the expression of the GPA1 protein, might be less active when cells are grown in galactose media. Since GPA1 is inhibiting signalling through the pheromone pathway when bound to the Ste4-Ste18(Gβγ)-complex, less GPA1 present would lead to more signalling through the pathway.
Finally, the ability of the modified GPA1 proteins to transduce a signal from a GPCR was investigated. While the results showed that the adenosine biosensor with wt GPA1 responded to the adenosine concentrations as expected, this response was not observed for any of the adenosine biosensors with modified GPA1 proteins. Thus, the modifications introduced a loss-of-function in respect to signal transduction in GPCR-based biosensors. The investigations as to whether the modifications introduced a gain-of-function in regards to signalling through cleavage by the TEV protease were inconclusive.