Team:UNILausanne/Engineering
Engineering success
Represillator system
The original repressilator system
The repressilator system is at the heart of our experiment – naturally this meant that we had to start our lab work by testing the original system. For this, we decided to adapt the “flask experiment” found in the paper by Potvin-Trottier et al. from 2016 to use in a plate reader, so that we could test multiple conditions and mutants at once.
Testing and redefining the protocol
To refine the flask experiment found in the source paper and adapt it to our own protocol, we had to test different growth media (EZ vs imaging media, found in protocols ), different ways of synchronising the oscillations (IPTG vs aTc) and different overnight incubation temperatures (ranging from 20°C to 37°C) in order to optimise the fluorescence readout.
One of the big problems we had early on was the formation of crystals in the bottom of the wells of our 96-well plate. To try and combat this we added Pluronic-F to our imaging media, a compound used for microfluidics to avoid this problem. However, we realised that this didn’t affect the results and so we continued using the media without Pluronic-F.


However, observing the results we realised that this didn’t affect any fluorescence measurements and so we continued using the media without Pluronic-F.
The plasmid choice journey
We started by using the plasmids pLPT119 and pLPT107 for the repressilator in E. coli Nissle 1917 Δclb (Part: BBa_K3482025) and pLPT41 as the sponge (Part: BBa_K3482026).
The plasmid pLPT107 was finally also not adapted, because of the degradation tag present at the end of the repressor genes. For this reason, we decided to replace it by the repressilator plasmid pLPT234 (Part: BBa_K3482023).
After a few tests, we also realised that CFP in pLPT119 was not acting as expected; this is when we discovered that in fact it’s under a constitutive promote (pRNA1), in this plasmid, and so doesn’t reflect the oscillations of the Tet repressor but ends up following the OD600.
We finally defined our various models with our chosen repressilator plasmids and prepared prediction models, to determine what we would expect to observe (See Figure 3 and 4.

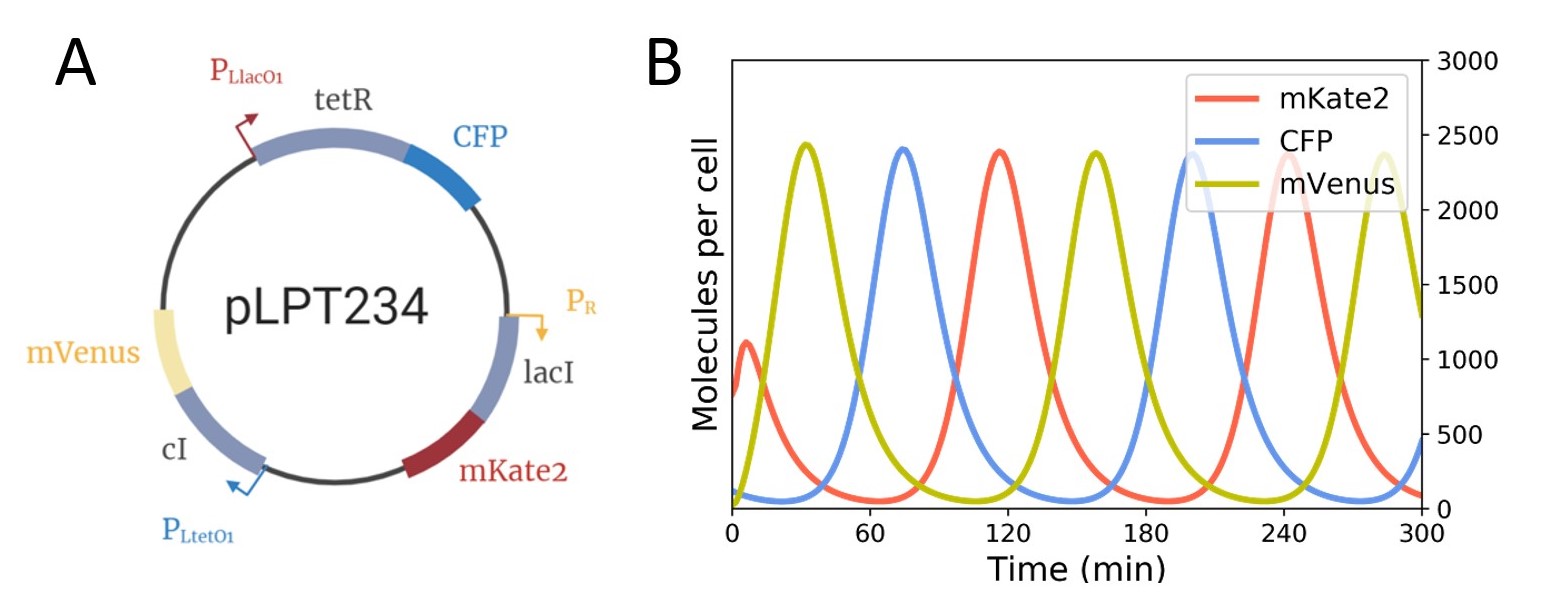
Further research into the system led us to use a triple sponge plasmid, pLPT145 (Part: BBa_K3482024), that led to similar oscillations as the pLPT41 sponge plasmid in E. coli DHL708. We therefore also tested these non-modified combinations, using this sponge plasmid with either pLPT119 or pLPT234 in E. coli DHL708, to figure out which one showed the most reliable oscillations. We obtained oscillatory patterns of expression in the repressilator plasmids we transformed in E. coli DHL708 for the pLPT119 plasmid and pLPT234.
In conclusion, we succeed to characterize and optimize 3 repressilator system combinations:
- pLPT234 + pLPT41
- pLPT234 + pLPT145
- pLPT119 + pLPT41
We were able to obtain clear and regular oscillations in these three systems, as we have shown in the experiment section ( See results on experiment section).
The modified repressilator system
Whilst one part of our wet lab team worked on optimizing the plate experiment protocol, another part was working on the assembly of the modified plasmids for the secretion of azurin and/or with a fluorescent reporter to test if everything worked as expected.
When we first started modifying our system, we originally wanted to use mKate2 as the fluorescent reporter on our sponge plasmid to track oscillations. However, we realised after the first few plate experiments that mKate2 detection was very unreliable in pLPT107 and pLPT234. This meant that we had to rethink our plans and we eventually decided on removing mVenus from the repressilator plasmid and adding it to the sponge plasmid.

mVenus removal and azurin integration
The removal of mVenus from either repressilator plasmid ended up being more difficult than expected. Manipulating such large fragments of DNA were the biggest challenge we faced, and we tried to overcome in multiple ways including:
- Modifying the PCR protocol for higher fidelity
- Separating the plasmid into multiple pieces to be stitched back together using Gibson assembly
- Site-directed mutagenesis to target mVenus expression
The main difficulty encountered here was the fact that the sequences were too similar, which led to the multiple difficulties to modify the sequence.
We managed to obtain the repressilator plasmid pLPT234 without mVenus and decided to test it without our modified sponge plasmids pLPT41 and pLPT145, containing each mVenus with or without azurin next to it.
Unfortunately, none of our systems gave clear fluorescence oscillations ( See results on experiment section).
We finally decided to test a last combination: the unmodified repressilator plasmid pLPT119 with the modified sponge plasmid pLPT41, that was containing the azurin gene. As we can see on Figure 6, this repressilator system gave us clear oscillations of mVenus, as we expected.

In conclusion, after many trials, tests and modifications, we managed to produce protocols, characterize and successfully transform the repressilator circuit into E. coli Nissle. We also managed to create a repressilatory system which was stable, while containing the azurin gene.
Azurin expression
The initial design
We then planned to evaluate the azurin expression module through three main steps: 1) purifying the azurin protein from E. coli BL21, a protein expression strain, 2) verify that the expressed protein is indeed azurin and 3) that azurin is secreted (found in the supernatant through the addition of a pelB-5D secretion tag).
Using Golden Gate assembly, we introduced azurin along with a TwinStrep His tag onto the pET22 backbone, under a T7 expression promoter. This construct was then transformed into E. coli BL21 which was left to incubate overnight and produce our protein in the presence of the correct inducer. However, our first attempt at purification showed that this would be more difficult than imagined. Even though we used a protein expression strain and plasmid, we did not observe any big overexpression bands in the gel. We did, however, see some slightly smaller bands which we were led to believe that it might be azurin, and so we decided to attempt a large-scale purification using this construct.
The large-scale purification didn’t yield any azurin and so we started exploring other ways of purifying this protein from the same strain.
We then tried placing our optimized azurin (with an 8-His tag for purification) downstream of a Pbad promoter in a different plasmid - this one could not be transformed into BL21 so we decided to try purification directly from E. coli Nissle 1917 Δclb. We also attempted the same thing using the azurin part established by the ETH Zürich team in 2017 (link part). These plasmids at first seemed promising during the small-scale purification, but once again we did not obtain any purified azurin when we attempted to scale up.
After these attempts, we then created several new constructs with azurin combined with a 3XFLAG Tag inserted into the pET22 expression plasmid under a T7 IPTG-inducible promoter. These were then once again transformed into E. coli BL21, which was left to grow in media containing IPTG. However, once again in the small-scale purification, we didn’t obtain any clear overexpression bands. This time, however, we looked closer at the parameters of our experiment and tried modulating the concentration of IPTG we were using to induce protein expression.

Figure 1A: SDS PAGE of bacterial lysate. Samples were induced for 4h with the mentioned IPTG concentrations. The OD600 of all cultures was normalized to 0.5. Samples were lysed for 10min at 95°C. and run on a 15% polyacrylamide gel for 30 minutes at 200V 1B: SDS-PAGE with the same culture conditions as the uninduced sample from Figure 1A. Purification performed using HisPur Ni-NTA Magnetic Beads, incubated for 5 seconds with our sample. Signaled in red is the expected azurin protein. Samples were run on a 15% polyacrylamide gel for 30 minutes at 200V.
We hypothesized that the overexpression of azurin may be toxic to this particular E. coli strain and thus tried the same with our chassis bacteria, E. coli Nissle 1917 Δclb. From this, we saw that the same effect occurred. Thus, we suggested that only the leaky expression of the T7 promoter gave a tolerable level of expression.
The second step
The SDS PAGE was not really representative of azurin expression because other proteins were often present at the same size of the anti-cancer peptide. So we designed azurin combined with a 3xFLAG tag at the N-terminus. We thus made the design choice to recombine a 3XFLAG flag at the N-terminus. This choice opened the ability to confirm both the secretion and identity of the azurin through proteomic analysis (Western blot and mass spectrometry) in E. coli Nissle 1917 Δclb. These were proven more adapted as they are sensitive detection methods.
During this analysis, we decided to test not one, but two secretion tags: pelB-5D and NSP4. Indeed, the found literature search gave additional choices that we decided to test in parallel to allow for a broader range of usage for future IGEM teams.
Summary
We made several adjustments throughout the module in response to our experiment results. One of which was the addition of a 3XFLAG flag tag at the N-terminus of azurin genes. In the end, these results lead us to conclude on the secretion and identity of azurin.
Kill switch system
Kill switch system
Introducing a genetically modified living organism into the body could cause some unexpected consequences, for example the bacteria invading the blood circulation, which can cause a septic shock. Moreover, GMO bacteria might pose a risk of unwanted escape and spread in the environment when they are secreted from the body. That is why we decided to add a biosafety and biocontainment mechanism known as a kill switch.
Our kill switch system allows the bacteria to stay alive only in the colon where we want them to fight the colorectal cancer. When the bacteria get in the outside environment, or into the blood circulation, the kill switch will kill the bacteria.
The kill switch system does this by sensing two environmental cues: 1) the temperature, and 2) the phosphate concentration.
In the colon the temperature is high and the phosphate concentration is low, and these represent the permissive condition of our kill switch. On the other hand, outside the body the temperature is lower than in the colon, and in the bloodstream the phosphate concentration is higher than in the colon, and therefore low temperature and high phosphate represent the non-permissive conditions of our kill switch (Figure 1). Moreover, the ingestion of an encapsulated pill containing phosphate allows it to also kill the bacteria on demand even in the colon.

To achieve this controlled cell death, we use toxin-antitoxin systems, where the cell is killed when the toxin is expressed in absence of the antitoxin. To incorporate the temperature logic, our design uses heat-inducible RNA thermosensor fused to the antitoxin genes, and heat-repressible RNA thermosensor fused toxin genes. The phosphate concentration logic is then incorporated by putting the antitoxin gene under the control of a phosphate-repressible promoter. The final design is depicted in Figure 2.
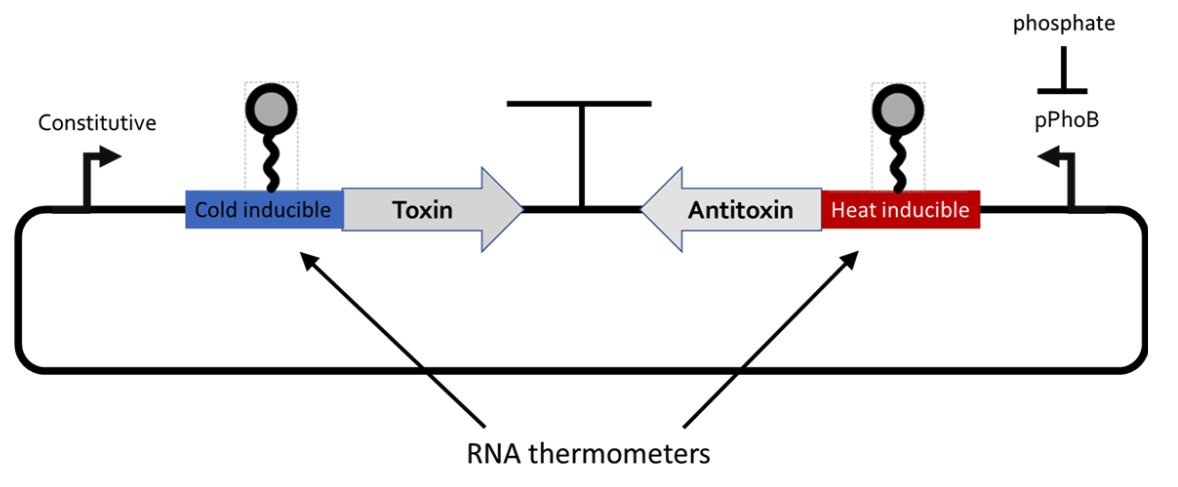
Since this design is a rather sophisticated and multi-responsive subject, we use a bottom-up approach where we first characterize the effect of every single part, and then gradually add more components onto our device. To increase our chances of success, we based our system on an already well-known toxin-antitoxin system in the literature. These are the toxin-antitoxin pair ccdB/ccdA (BBa_K3482016) and miniColicin E2/IM2 (BBa_K3482017). We then chose a very promising heat-inducible and heat-repressible RNA thermosensor also from the literature [1] [2]. The core promoter of a well-characterized phosphate-repressible promoter was taken from the iGEM Registry. Here, we further describe our development of the plasmid system based on miniColicin E2/IM2.
As we did not know exactly which of the two promoters, pTet and pTac, was the one with the highest leaky/basal expression, we designed our Gibson assembly such that the toxin and antitoxin genes could insert under either of promoters.

When we sequenced our cloning strain transformants we observed that the ones that inserted the toxin under pTet mostly showed mutations in the toxin gene (data not shown). We then reperformed the transformation by adding either aTc or IPTG to the transformant media, and were able to select this way under which promoter the antitoxin would be inserted (Figure 3). As the first sequencing of the first transformants showed no mutations when the antitoxin was inserted under pTet and the toxin under pTac, we continued working with this configuration. This first kill switch plasmid is pKC1 (Figure 3A).

Figure 4: Bottom-up plasmid design to test the functionality of the kill switch components. a) pAND is the empty vector containing pTac and pTet promoter without any gene. b) pKC1 contains the miniColicin E2 toxin under pTac and the IM2 antitoxin under pTet. c) pKC3 contains the heat-inducible thermosensor fused to the antitoxin gene.
To perform a first qualitative assessment of the functionality of the kill switch responsiveness towards induction of aTc and IPTG induction, we plated a lawn of E. coli Nissle 1917 and added orthogonally placed cardboard strips soaked with aTc and IPTG (Figure 5). The plate showed that close to the IPTG strip very little cells grew, and that further away from it the diffusion of aTc was able to sufficiently induce the antitoxin and rescue the cell growth.

We then performed a quantitative assessment of the effect of the stimulation of toxin and antitoxin on the growth of our E. coli Nissle 1917 strain by adding different amounts of aTc and IPTG inducer concentrations, and measuring growth curves in the plate reader (Figure 6).
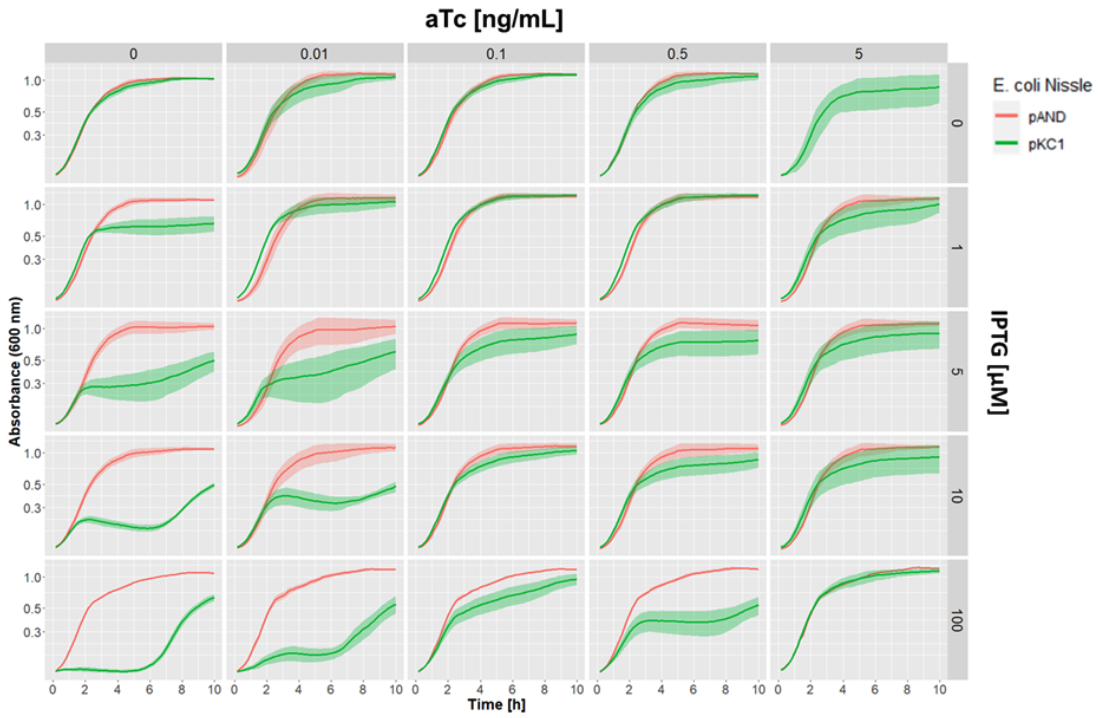
This allowed us to clearly determine the different ranges of aTc and IPTG concentration that give different growth inhibition profiles.
Finally, we then inserted the heat-inducible RNA thermosensor upstream of the antitoxin genes in the plasmid pKC3 (BBa_K3482017). In order to prove that the heat-inducible thermosensor reduces the translation of antitoxin at low temperature and therefore the viability of the cells, we grew the strain at different temperatures reflecting the temperature inside (37°C) and outside (≤25°C) the body, respectively (Figure 7).

At permissive temperature (37°C) the pKC3 containing cells grew a bit slower than the two controls, but their viability was not impaired. Indeed, after 10 hours the cells containing either pAND or pKC1 seemed to reach a plateau where the pKC3 cells were still growing. At lower temperature, the viability of pAND and pKC1 cells remains unchanged while pKC3 viability was significantly impaired.
By doing this we proved that the plasmid pKC3 containing the part BBa_K3482017 is able to control the viability of its host.
References
[1] Hoynes-O’Connor, A., Hinman, K., Kirchner, L., & Moon, T. S. (2015). De novodesign of heat-repressible RNA thermosensors in E. Coli. Nucleic Acids Research, 43(12), 6166–6179.
[2] Sen, S., Apurva, D., Satija, R., Siegal, D., & Murray, R. M. (2017). Design of a Toolbox of RNA Thermometers. ACS Synthetic Biology, 6(8), 1461–1470.