Team:UNILausanne/Experiments
Experiments
Represillator system
Background
Treatments for colorectal cancer such as chemotherapy, surgery and radiotherapy are associated with severe side effects and are limited in effectiveness. We aim to implement a system that can produce an anticancer drug called azurin in a circadian rhythm. For this purpose, we set to use the repressilator.
The repressilator is a synthetic regulatory network of three genes encoded on a plasmid. These three genes repress each other in a feedback loop. As a result, the genes are cyclically expressed one after the other at regular intervals over time, which allows us to produce our anticancer drug at regular intervals. For our system to work efficiently, a second plasmid called a sponge plasmid is applied together with the repressilator. This sponge plasmid contains additional repressor binding sites which allows for the “absorption” of noise and makes the oscillations more regular and robust.
Repressilator plasmids
For our experiment, we want to test two types of repressilator plasmids:
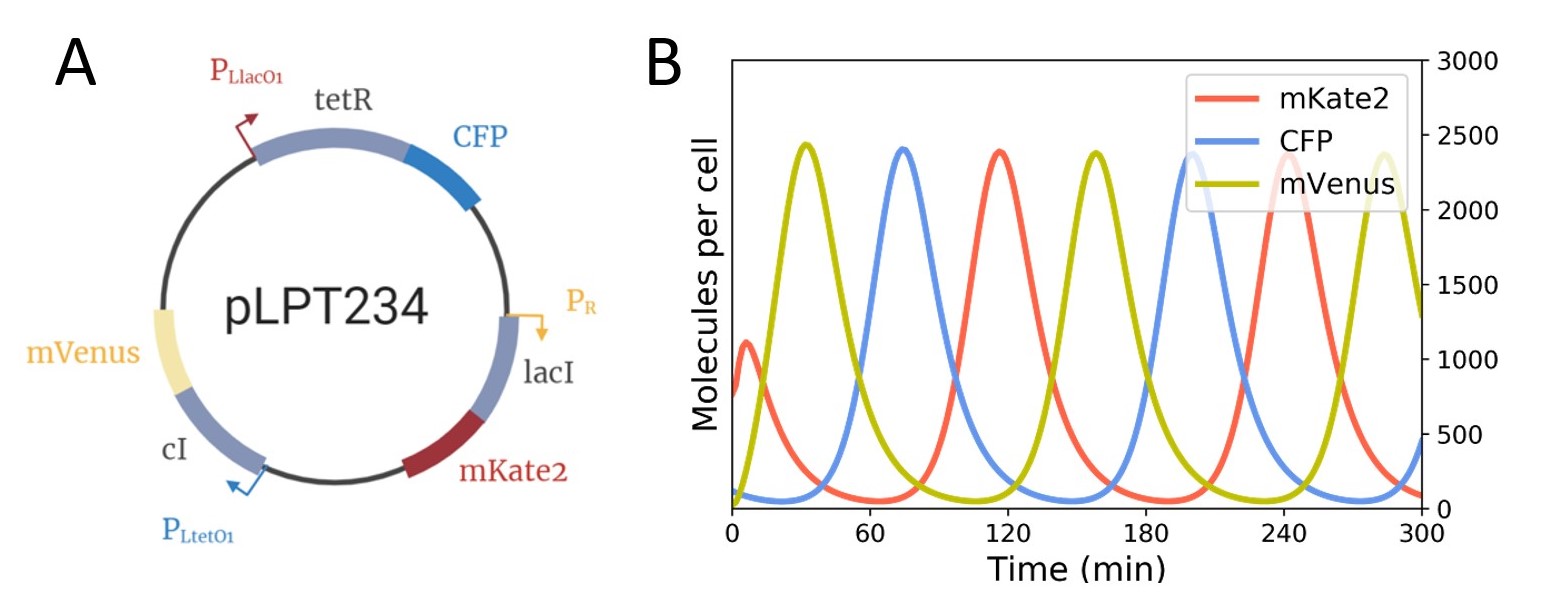
The first one is pLPT234 [BBa_K3482023] (Figure 1A). It is composed of three repressors (tetR, lacI and cI), that are under the regulation of three distinct promoters (PLlacO1, PRNA1and PLtetO1 respectively). The oscillations can be followed thanks to three fluorescent reporters (CFP, mKate2 or mVenus) which are under the control of the same three promoters. The three repressors are connected in a circuit, where one repressor inhibits the next. This feedback loop circuit leads to the oscillatory expression of three colors (Figure 1B).
The second repressilator plasmid is pLPT119 [BBa_K3482025] (Figure 2A). The composition of the plasmid is similar to the plasmid pLPT234 [BBa_K3482023]. Here there is only the fluorescent reporter gene mVenus associated with a repressor protein (λ), with CFP on the plasmid but under a constitutive promoter. All three repressor proteins are still present, and the system still functions as in pLPT234 BBa_K3482023], but we can only follow the oscillations using mVenus. (Figure 2B).

Sponge plasmids
In order to have robust and regular oscillations, the repressilator plasmid needs to be combined with a so-called “sponge” plasmid. This plasmid contains one or three promoters (including the repressor binding sites) already present in the repressilator plasmids. In our project, we worked with two different sponge plasmids (Figure 3). The first plasmid is pLPT41 [BBa_K3482026] (Figure 3A) and contains a PLtetO1 promoter. The second plasmid is pLPT145 [BBa_K3482024] (Figure 3B) and contains the three promoters present in the repressilator plasmid pLPT234 BBa_K3482023]. We tested these two sponge plasmids in combination with our two repressilator plasmids, to determine which combination resulted in the best oscillations.

All of the plasmids presented here were described in Potvin-Trottier 2016. They were obtained and ordered from Addgene. They were made accessible by Prof Johan Paulson [1]. We decided to base our experiment on these plasmids and modify the system afterwards for our project.
Procedure
Strains
For our repressilator experiments, we worked with two different strains.
Experiment goal
We had two main goals in this experiment:
First we wanted to determine if our repressilator system worked correctly in our two strains (E. coli DHL708 and E. coli Nissle Δclb) and if we could detect them without using microfluidics. To achieve this, we tested the following combinations in various conditions to optimize our protocols:
Our second goal was to integrate our anticancer drug (azurin) in our repressilator plasmid. We then needed to determine if the oscillations of our gene circuit were still stable after the integration of azurin. We tested the various following modifications:

Experimental procedure
Overnight cultures of E. coli DHL708 or E. coli Nissle Δclb with one of the combinations of repressilator and sponge plasmids listed above were prepared in imaging media ( See protocol). with the corresponding antibiotics and the synchronizing molecule anhydrotetRacycline (aTc, 0.5 μL/mL). The imaging media is a growth media with a very low background fluorescence. ATc is a molecule that inhibits the tetR repressor; upon addition of aTc to the culture, all bacteria containing a repressilator plasmid halt oscillations at the same time point. After removal of aTc, all the bacteria start oscillations simultaneously and thus the incubation with aTc effectively synchronizes the oscillations in a bacterial population.
In the morning, we resuspend the cells in fresh imaging media without aTc and transferred them to a 96-well plate. We diluted the cells to reach an optical density at 600 nm (= OD600) of 0.2 and put the plate in a plate reader at 37°C and 180 rpm. Every hour, we checked the OD600 and diluted the cells back to an OD600 of 0.2, to keep the cells in exponential phase. This was important, because the repressilators do not produce oscillations when cells are in a stationary phase. Fluorescence measurements were taken every hour for approximately 10 hours.
Results
Overview
First, we will present the results of the original repressilator plasmid pLPT234 [BBa_K3482023] in combination with the sponge plasmids: pLPT41 [BBa_K3482026] or pLPT145 [BBa_K3482024]. We then compared the results of the modified version of pLPT234 [BBa_K3482023] (without mVenus) with the modified sponge plasmids (with mVenus and with or without azurin). We will then present the results of the original plasmid pLPT119 [BBa_K3482025] in combination with the sponge plasmid pLPT41 [BBa_K3482026] containing either the azurin gene or a fluorescence reporter gene. Finally, we will compare this system with pLPT119 [BBa_K3482025] in combination with our modified sponge plasmid in which the azurin gene has been integrated.
First part: Repressilator plasmid pLPT234 [BBa_K3482023]
We tested our various repressilator systems without any modifications, starting with the combination of repressilator plasmid pLPT234 [BBa_K3482023] and sponge plasmid pLPT41 [BBa_K3482026] and pLPT145 [BBa_K3482024] in E. coli DHL708. In Figure 5, we show the fluorescence over time after the synchronization of the bacterial culture.


The combination of the repressilator plasmid pLPT234 [BBa_K3482023] with the sponge plasmid pLPT145 [BBa_K3482024] (Figure 6) gave clear and stable oscillations that correspond to our expectations and our model (Figure 1B, See modeling page. ). The second combination, the plasmid pLPT234 [BBa_K3482023] and pLPT41 [BBa_K3482026], gave less clear and stable oscillations (Figure 5). These results could be explained by the fact that the sponge plasmid pLPT41 [BBa_K3482026] only has one promoter of the repressilator plasmid, compared to the sponge plasmid pLPT145 [BBa_K3482024] that has all three. This sponge plasmid pLPT41 [BBa_K3482026] seems to compensate for the noise of the repressilator plasmid less than pLPT145 [BBa_K3482024] due to the lower number of promoters.
Since we demonstrated that the repressilator system is functional, we decided to test the repressilator in combination with a sponge plasmid containing the azurin gene, which should show oscillations of azurin (Figure 4B, C and D). The systems were integrated in our probiotic strain E. coli Nissle 1917 Δclb.



We tested our modified sponge plasmids with or without azurin linked to the fluorescence gene mVenus separately. In Figures 7 and 8, we can see that no clear oscillations are visible. It might be possible that the shift of the mVenus reporter from the repressilator plasmid to the sponge plasmids disturbed our oscillations in this assembly. Indeed, the sponge plasmid has a higher copy number compared to the repressilator plasmid (~20 compared to ~5). The shift of the fluorescent reporter mVenus from one plasmid to another might perpetuate the dynamical equilibrium of the system and even cause burden. Another explanation would be that the repressilator system, and synthetic biology in general, is really dependent on the context. Modifying even slightly some synthetic systems might completely disturb the system and the results obtained [2].
In parallel, we tested the expression behavior of azurin when integrated in the sponge plasmid. In Figure 9, we can observe again that we also obtained noisy and unstable fluorescence signals. We were therefore not able to obtain clear oscillations with our modified repressilator and thus couldn’t observe the oscillatory behavior of azurin. Various explanations to these results can be given. First, the shift of the reporter gene could have perturbed the repressilator system, as it seems to be the case in Figures 7 and 8. It is also possible that the integration of azurin disturbed the oscillations. However, the results from Figure 12 seem as noisy as the results obtained with the construct without azurin. In conclusion, it is not possible to determine with this construct if the integration of azurin is negatively impacting the oscillations.
Second part: Repressilator plasmid pLPT119 [BBa_K3482025]
After observing that we were not being able to modify the three reporter repressilator plasmid pLPT234 [BBa_K3482023] without impacting the oscillations, we decided to test the simpler version of the repressilator (pLPT119). We then tested again the repressilator plasmid without any modification in combination with the sponge plasmid pLPT41 [BBa_K3482026] in E. coli DHL708. In Figure 5, we show the fluorescence over time after synchronization of the bacterial culture.

In Figure 10, the decrease and following increase of mVenus fluorescence over time is clearly visible, which is consistent with our model ( See modeling page. ). The CFP fluorescence signal is stable for the first 9 hours, as we expected. However after this period, its fluorescence slowly started to decrease over time. This indicates that the repressilator begins to be unstable after approximately 10 hours of oscillations. This problem could be resolved by resynchronizing the cells with aTc. Looking at the fact that our experiment lasted between 10 and 12 hours, these results are still confirming that our repressilator system was working correctly for the maximum period of time where we would take measurements.
After confirming that our repressilator system containing the repressilator plasmid pLPT119 [BBa_K3482025] in combination with the sponge plasmid pLPT41 [BBa_K3482026] works for at least 10 hours, we decided to test our modified system, presented in Figure 4A. This time, the plasmids were transformed in E. coli Nissle Δclb. As before, we expected the fluorescent reporter mVenus to oscillate with time, and CFP levels to be stable.

In Figure 11, we can observe the results of this new repressilator system. As before, we see that the fluorescence of mVenus is oscillates over time. During our experiment, we observed that the cells were also growing a bit faster, which explains the shift of oscillations observed compared to the results of Figure 5. The fluorescence of CFP is however increasing with time, and then stabilizes for the 3 last hours of measurements. This result is a bit surprising and does not correspond exactly to our expectations. It is possible that the presence of azurin or the faster growth of the cells at the beginning of the experiment influenced the production of CFP during the first hours and that the stable state was reached with a delay.
In summary, these results suggest that the pLPT119 [BBa_K3482025] repressilator and pLPT41 [BBa_K3482026] sponge plasmid work in both E. coli DHL708 and E. coli Nissle 1917 Δclb. The oscillation is stable until around 10 hours after synchronization. While the growth speed of E. coli Nissle 1917 Δclb is slightly increased, the oscillation period is not affected by the possible expression of azurin in the sponge plasmid.
Conclusion
With the repressilator experiments, we could confirm that we were able to obtain oscillations with all of our unmodified repressilator plasmids, integrated in E. coli DHL708. We were however not able to characterise the oscillatory behaviour of the azurin expression in the modified system with the repressilator plasmid pLPT234 [BBa_K3482023], because the shift of the fluorescent reporter gene mVenus disturbed the oscillation system. In the modified repressilator system based on the plasmid pLPT119 [BBa_K3482025], we were able to demonstrate that transferring our modified repressilator system gave to E. coli Nissle 1917 Δclb yielded similar results as to what was observed in the cloning strain. We could then confirm that the repressilator system is working in our probiotic and that the integration of azurin is not disturbing the oscillations. To go further in this experiment, we should integrate an independent fluorescence reporter link to azurin, to demonstrate that the azurin is following an oscillation behavior.
References
- Addgene: Johan Paulsson Lab Plasmids. https://www.addgene.org/Johan_Paulsson/.
- Kwok, R. Five hard truths for synthetic biology. Nature vol. 463 288–290 (2010).
Azurin expression
Introduction
With the intent to propose an anti-cancer therapeutic agent that is producible in an Escherichia coli strain and that poses no strong side effect to the patient, we found azurin as a potential anti-cancer drug. Azurin is a blue copper-binding redox protein originally found in the periplasmic membrane of Pseudomonas aeruginosa[1]. It has recently been discovered to have an anti-cancer effect by targeting the well-known p53 apoptosis pathway and by preferentially entering cancerous cells [2]. Because we are using E. coli Nissle 1917Δclb as our chassis organism and designed our azurin to be secreted, we codon-optimized the azurin gene based on E. coli (ATCC_8739_uid58783) using the “Genome calligrapher” from ChristenLab at the ETH Zürich [3]. Because azurin is a periplasm membrane protein, it is not normally secreted by bacteria. To reach our goal of secreting azurin, we added a secretion tag (pelB-5D) to the C-terminus of our sequence, which originates from a strain of Erwinia carotovora [4], [5].
Overview
Our objective is to prove the expression and secretion of our optimized azurin in an oscillatory manner. Furthermore, we wanted to prove the anti-cancer effectiveness by testing it using a cell viability assay on cancerous cells. We first performed a purification of azurin, then we characterized its expression by SDS PAGE, Western Blot, and Mass Spectrometry (MS). Lastly, we applied our construct on a colorectal cancer model cell line to assess its cytotoxicity.learn more about this in our proof of concept
Azurin purification
To explore the therapeutic effect of azurin on cancer cells and to be able to compare it to our design, we needed to express and purify our protein. Multiple concentrations of inducing molecules, IPTG, were tested. The SDS PAGE was used to isolate and show the presence or absence of azurin. For this, we used the expression strain E. coli BL21 transformed with the expression vector pET22 (BBa_3482044), containing the optimized azurin coding gene mentioned above with a 6-His tag. We induced the expression using 50 µM or 500 µM of IPTG for 4 h (Figure 1A). In a second step, we selectively purified our azurin protein using the 6-His tag in combination with His Mag Sepharose® Ni (Sigma GE28-9673-90) (Figure 1B).

We can observe in Figure 1A the presence of three bands around 15 kDa, which is the size range of our azurin (14 kDa). Interestingly, the uninduced sample shows the highest intensity and we hypothesized that this could be due to intrinsic toxicity upon overexpression using IPTG. Thus, the leaky expression of the T7 promoter may allow for a higher yield of our protein expression. Upon purification using the specific 6-His tag, we can see in Figure 1B only one band (red box), which corresponds to the high-intensity band previously observed. From these experiments, we concluded that the purification of azurin was successful.
Proof of expression of azurin
To verify the expression of the azurin that we successfully cloned into our sponge plasmid and purified, we concatenated a widely used and sensitive 3XFLAG tag at the C-terminus of our azurin gene (BBa_3482040). This allowed us to perform a Western blot, which allowed us to confirm the presence or absence of our expressed azurin. In addition, we modified the existing part BBa_K2500001 (truncated azurin uploaded by the ETH Zürich team of 2017) with either the secretion tag pelB-5D or NSP4[6] and investigated its secretion capability.
First, we tested our chassis, E.coli Nissle 1917Δclb, which has been transformed with a modified sponge plasmid (usable in combination with a repressilator plasmid in the future) which contains our target genes constitutively expressed under a pTet promoter (BBa_K3482040, BBa_K3482041, BBa_K3482038, and BBa_K3482037). We then performed a Western blot with cultures set to an OD600 of 2.0. The culture supernatants, separated from bacterial pellets by centrifugation and remaining bacterial pellet lysate were analyzed (Figure 2).

Samples are as follow: 1 = azurin-3XFLAG; 2 = truncated azurin-3XFLAG; 3 = pelB-5D-truncated azurin-3XFLAG; 4 = NSP4-truncated azurin-3XFLAG.
We can observe the presence of the azurin linked to the FLAG tag in all cell lysate extracts. In addition, we can see that the full-length azurin as well as both versions with a secretion tag (pelB or NSP4)(Figure 2, samples 3 and 4) are present in the supernatant. Importantly,we can see that no secretion occurred without a secretion tag (Figure 2, sample 2) in comparison with the full azurin protein(Figure 2, sample 1). This leads us to conclude that our azurin is indeed expressed and secreted. In addition, we successfully added the secretion aspect to an already characterized azurin part (BBa_2500001).
However, due to the presence of smearing and unsharp bands, we wanted to further test the proof of expression of azurin by E. coli Nissle 1917Δclb. To this end, we referred to the experts in the field of protein analysis: The Protein Facility Center at the University of Lausanne (PAF UNIL).
With their expertise in immunoblotting and mass spectrometry, we were able to further assess our preliminary results. The same samples as in the previous experiments were analyzed. SDS PAGE (Figure 3) was performed first to separate proteins by size, then upon analysis, two bands corresponding to the size of azurin were cut and further characterized using mass spectrometry (Figure 4):

We can observe multiple bands at the predicted size range of azurin, but no clear overrepresented band (Figure 3). Because of this, two bands of different size range A and B were cut and analyzed using Mass spectrometry (Figure 4).
Note that due to the absence of visible bands on SDS PAGE (data not shown) of the supernatant, no MS analysis was done for the supernatant. This does not mean, however, that no protein can be found in the supernatant, thus secreted, but rather that the SDS PAGE methods, in contrast with western blot (Figure 2, supernatant sample) are probably not sensitive enough to be able to see the azurin secretion.
Using MS allowed us to assess the nature of the protein content of our cell lysate as well as being able to characterize the relative quantification of the constitutive expression of our construct. Indeed, with a sequence coverage of 97.7% (difference due to the presence of truncated azurin as well as secretion and/or flag tag) based on the BBa_3482022 sequence, it is possible to confirm that the purified, as well as the recombined protein with 3XFLAG, are azurin.

We can see that the presence of azurin was detected only in band A Figure 3), which was of higher size than expected. This can be due to the differentiation of migration occurring during SDS PAGE and due to conformational change which can occur when tags and flag tags are concatenated to a protein. We can also observe that the relative amount of azurin is lower for the truncated azurin containing a secretion tag. We hypothesis that due to secretion, we found less azurin in the cell lysate.
In summary, we were able to detect the presence of azurin and quantify the relative amount of protein constitutively expressed under the pTet promoter in E.coli Nissle 1917Δclb for the BBa_K3482040, BBa_K3482038, and BBa_K3482037 part.
Kill switch system
Achievements
- Contribution (bronze medal): we provide the first and extensive characterization of the toxin-antitoxin systems ccdB/ccdA (BBa_P1010, BBa_K1075032) and miniColicin E2/IM2 (BBa_K1976048, BBa_K1976027) and identified the promoter induction range for differential cell growth inhibition. We also demonstrate that the widely used E. coli Nissle 1917ΔclbA has a ccdB sensitive gyrase genotype
- Engineering success (silver medal): we characterized our new composite part (BBa_K3482017)
- Proof of concept (gold medal): we demonstrate that our kill switch system controls cell growth effectively depending on temperature.
Introduction
The aim of the kill switch is to constrain the activity of our engineered probiotic Escherichia coli Nissle 1917 Δclb to the (permissive) colonic environment. The viability of the cells should be reduced under non-permissive conditions where either the temperature is decreased or the phosphate concentration is increased (Table 1). The goal is to develop a system that on one hand kills the bacteria when they get in the environment where it’s colder (at least in the waste water), and on the other to kill the bacteria if they were to enter the bloodstream, where higher phosphate concentrations are present in comparison to the colon. This would also allow to remove the bacteria from the patient’s gut by providing an encapsulated pill which would release high amounts of phosphate in the colon.
Table 1 Kill switch viability logic with temperature and phosphate concentration as inputs.
| Condition | Temperature [°C] | Phosphate concentration | Predicted viability of host cell |
| Permissive | 37 | Low | High |
| Non-permissive | 37 | High | Low |
| Non-permissive | 25 | Low | Very low |
We employ two toxin-antitoxin plasmid systems based on toxin-antitoxin pairs ccdB/ccdA (BBa_P1010, BBa_K1075032) and miniColicin E2/IM2 (BBa_K1976048, BBa_K1976027). The ccdB toxin is a DNA gyrase inhibitor which, in absence of the ccdA antitoxin, leads to DNA breakage and cell death [1]. This ccdB toxin is active only against bacteria because DNA gyrase is only present in bacteria and lower eukaryotes [2]. The miniColicin E2 toxin is a DNA endonuclease which, in absence of the IM2 antitoxin, also leads to cell death [3]. The miniColicin E2 gene does not code for neither the translocation nor receptor binding domain, and can therefore not be secreted by the bacteria. In case the toxin were released in the gut environment, it would be digested by the highly abundant proteases [4]. Therefore, these toxins do not pose any health risk.
The toxin and antitoxin genes were cloned into pAND-MSC (BBa_K3482021), a plasmid that allows for inducible expression of the genes cloned under the pTet and pTac promoters, with IPTG and aTc respectively [5]. The toxins were inserted under the control of pTac, whereas the antitoxin under that of pTet.
To incorporate the temperature logic, the heat-inducible 2U RNA thermosensor (BBa_K3482000) was inserted upstream of the antitoxin genes, while the F2 heat-repressible RNA thermosensor (BBa_K3482001) can be inserted upstream of the toxin genes. The heat-inducible RNA thermosensor reduces mRNA translation efficiency at lower temperature by forming a hairpin structure that conceals the ribosome binding site (RBS) [6]. Conversely, the heat-repressible RNA thermosensor reduces the mRNA translation efficiency by exposing a RNase E cleavage site, which is concealed at lower temperature by an hairpin structure [7]. Lastly, to incorporate the phosphate concentration logic, the pTet promoter can be replaced by a phosphate-repressible pPhoB promoter (BBa_K116401).
Our kill switch design is based on two toxin-antitoxin systems that enable us to guarantee plasmid retention of our repressilator and sponge plasmids by establishing a so-called plasmid addiction system. This is a tool based on the post-segregational killing mechanism [8]. This is an established system used to maintain plasmids without using antibiotics. It relies on the higher stability of the toxin compared to the antitoxin. This means that daughter cells who do not inherit the plasmid are killed by the remaining toxin in the cell, while the antitoxin, less stable, is degraded . Therefore, the kill switch module coding for toxin and antitoxin can be used to replace the antibiotic resistance genes of our repressilator and sponge plasmid. Consequently, our therapeutic strain will be even safer since it cannot transfer any antibiotic resistance genes to the other bacteria of the gut microbiota.
Objectives
The objectives of the experiment are the following:
- Study the cell-growth effect of different concentrations of antitoxin and toxin inducers, aTc and IPTG, respectively.
- Assess the effect of the 2U RNA thermosensor on the expression of antitoxin at 37°C and 25°C.
Protocol
Bacterial strains
We use E. coli Nissle 1917 ΔclbA [9]. The clbA gene is part of an endogenous cluster of genes coding for the biosynthesis of hybrid nonribosomal peptide-polyketide(s), which confer the strain the ability to induce double strand breaks in eukaryotic cells. Thus, the inactivation of the clbA gene abrogates the genotoxic ability strain. E. coli Nissle 1917 ΔclbA has been transformed with the plasmids listed in Table 2 and depicted in Figure 1.
Table 2 Plasmids used for the kill switch experiments.
| Plasmid name | Backbone | Toxin | Antitoxin | RNA thermosensor name, function | Reference |
| pAND | pAND-MSC(BBa_K3482021) | - | - | - | [5] |
| pKA1 | pAND-MCS (BBa_K3482021) | ccdB(BBa_P1010) | ccdA(BBa_K1075032) | |
|
| pKC1 | pAND-MCS (BBa_K3482021) | miniColicin E2(BBa_K1976048) | IM2(BBa_K1976027) | |
|
| pKC3 | pAND-MCS (BBa_K3482021) | miniColicin E2(BBa_K1976048 ) | IM2(BBa_K1976027) | 2U, heat-inducible antitoxin expression(BBa_K3482017) | [6] |

For the experiments, the following strains were used:
- Escherichia coli Nissle 1917 ΔclbA transformed with pKA1 or pKC1 harboring toxin and antitoxin genes under the control of pTet (BBa_K3482012) and pTac (BBa_K3482011), respectively.
- Escherichia coli Nissle 1917 ΔclbA transformed with pAND. Negative control for the effect of the toxin.
- Escherichia coli Nissle 1917 ΔclbA transformed with pKC3 harboring miniColicin E2 toxin and heat-inducible IM2 antitoxin.
Material
The required solutions are listed in Table 3. Further material needed is:
- 14-mL round bottom culture tubes with breathable cap
- Biotek Synergy h2, Plate reader
- Nanodrop
Table 3 Solutions used for the kill switch experiments.
| Solutions | Stock concentration |
| Luria Broth (LB) | |
| aTc stock solution | 100 ng/mL |
| IPTG stock solution | 1 M |
| Kanamycin | 50 mg/mL |
Procedure
- Pick single colonies from plates to inoculate 1 mL LB with 50 µg/mL kanamycin in tubes.
- Incubate the culture at 37°C, shaking at 200 rpm for 3-6 h.
- Measure the OD600 and dilute the cultures to OD600 = 0.05 in 2 mL LB with 50 µg/mL kanamycin.
- Inoculate the 96-wells plate with 90 µL of the OD600 = 0.05 culture according to the chosen plating layout.
- Prepare dilutions of aTc and IPTG in LB with 50 µg/mL kanamycin.
- Add 10 µL of the 10X aTc of the desired final concentration to the appropriate wells.
- Add 10 µL of the 10X IPTG of the desired final concentration to the appropriate wells.
- Incubate the plate at either 37°C or 25°C under continuous shaking in the plate reader for 10 h or overnight. Set an absorbance measurement at 600 nm every 10 minutes.
Data analysis
Raw data was exported using the automatic export function of Gen5 (version 5.3). These raw data were processed in Excel and analyzed in R.
Results
ccdB/ccdA kill switch plasmid pKA1
We first tested our pKA1 plasmid encoding for the ccdB toxin and ccdA antitoxin in E. coli Nissle 1917 ΔclbA. We used different concentrations of IPTG and aTc to study the effect of differential expression of the toxin and antitoxin on the growth ofour strain. As a control, we used the strain transformed with the empty vector pAND (Figure 2).
We observed desired growth inhibition of the strain with pKA1 at high IPTG and low aTc concentrations. The pAND strain showed as expected no alteration in growth in any of the tested conditions. The uniformity of these growth curves of the strain with pAND demonstrates that the standardized culturing and OD normalization of our protocol can yield reproducible results.
The sensitivity towards ccdB demonstrates that E. coli Nissle 1917 has a sensitive gyrase genotype, contrasting other E. coli strains such as DH5α which has a gyrA96 genotype which renders them resistant to the toxin [10]. Interestingly, cell growth rescued by expression of ccdA was only noticeable in the transition from 0 to 0.1 ng/mL and from 10 to 50 ng/mL aTc induction.
We saw an effect of IPTG on growth inhibition starting from 5 µM IPTG. At maximum IPTG concentration, we observed complete absence of growth of the pKA1 strain for the first 7 h. However, afterwards, we observed that the bacteria were able to grow. This was probably due to mutations in the kill switch system. In contrast, at lower IPTG concentrations (5 and 10 µM), we still observed an inhibitor effect on growth, which albeit being less strong, was more constant and persisted for at least 10 h. This suggests that decreasing the expression of the toxin lowers the selective pressure on the kill switch system, which makes this configuration more evolutionarily stable.
Overall, we demonstrated that the ccdB/ccdA encoding pKA1 plasmid could efficiently inhibit the growth of E. coli Nissle 1917 at 100 µM IPTG for up to 7 h, and provided even longer growth inhibition at 5 and 10 µM IPTG.

MiniColicin E2/IM2 kills switch plasmid pKC1
Secondly, we tested our pKC1 plasmid encoding for the miniColicin E2 toxin and IM2 antitoxin in E. coli Nissle 1917 ΔclbA. Again, we used different concentrations of IPTG and aTc to study the effect of differential expression of the toxin and antitoxin on the growth ofour strain. As a control, we used the strain transformed with the empty vector pAND (Figure 3).
As with E. coli Nissle 1917 with pKA1, we observed desired growth inhibition of the strain with pKC1 at high IPTG and low aTc concentrations. Again, as expected the pAND strain showed no alteration in growth in any of the tested conditions. Contrastingly to E. coli Nissle 1917 transformed with pKA1, the induction of aTc was clearly able to rescue cell growth. For increasing concentrations of IPTG, also increasing concentrations of aTc were necessary to rescue cell growth. This linear relationship was compatible with the experiments demonstrating binding of IM2 to Colicin E2 with a 1:1 stoichiometry [11]. Similar to E. coli Nissle 1917 pKA1, cell growth could be completely inhibited for 6-8 h (IPTG: 100 µM, aTc: 0 ng/mL) before observing rapid growth. Also similarly to pKA1, the reduction in IPTG concentration gave a lesser but more constant growth inhibition, which also persisted to at least 10 h.
Overall, we demonstrated that the miniColicin E2/IM2 encoding pKC1 plasmid could efficiently inhibit the growth of E. coli Nissle 1917 at 100 µM IPTG for up to 6 h and provided even longer growth inhibition at 5 µM IPTG.

MiniColicin E2/ heat-inducible IM2 kill switch plasmid pKC3
Finally we tested our pKC3 plasmid encoding for the miniColicin E2 toxin and heat-inducible IM2 antitoxin in E. coli Nissle 1917 ΔclbA. Again, we used different concentrations of IPTG and aTc to study the effect of differential expression of the toxin and antitoxin on the growth ofour strain. As a control, we used the strain transformed with the empty vector pAND. The comparison of the pKC3 with the the pKC1 strain (no thermosensor), allowed to determine the effect of the growth inhibition caused by the thermosensor (Figure 4).
Our desired outcome was to observe growth of the strain with pKC3 at 37°C but not at 25°C for a given combination of aTc and IPTG concentration. This behaviour was clearly observed for the low aTc and IPTG concentrations, while the strain with pKC1 (no thermosensor) did not show any growth impairment at 25°C. While it might seem surprising that the effect of the toxin is obtained also without addition of IPTG, it has to be considered that inducible promoters have a baseline (leaky) expression even when no inducer is added.Our results suggest that the leaky expression of the pTet and pTac promoter yielded the best balance of toxin and antitoxin expression for the efficient growth inhibition at low temperatures. The growth inhibition of the strain with pKC3 at 25°C could be clearly rescued by the addition of aTc. It can also be noticed that at 37°C and in absence of aTc, the pKC3 strain showed a slight growth delay compared to the strain with pKC1.This is due to the fact that even at 37°C the thermosensor slightly reduces the translation efficiency of the gene, which it is coupled to [6].
In comparison to pKA1 and pKC1 plasmids, the growth inhibiting effect of pKC3 at 25°C seems to last longer, and for the vast majority of aTc and IPTG concentration combinations no sharp increase in growth was observed at any measured time point. This might be a result of the decrease in growth rate at lower temperature, which might also aid in reducing the selective pressure on the cells to mutate the components of the kill switch.
Overall, we demonstrated that the miniColicin E2/ heat-inducible IM2 encoding pKC3 plasmid could effectively inhibit the growth of E. coli Nissle 1917 at 25°C for low aTc and IPTG concentrations.

Conclusion and outlook
Impact of observed growth inhibition on the feasibility of our design for clinical applications
We demonstrated that our toxin-antitoxin plasmid systems enable us to efficiently control cell viability. Doing so, we performed extensive measurements on the ccdB/ccdA and miniColicin E2/IM2 parts, which were previously uncharacterized on the iGEM Part Registry. Moreover, we provide proof of concept for a temperature sensitive kill switch, which allows to stop cell growth under the non-permissive condition of low temperature.
The main threat to our system are mutations of the kill switch components, as this can cause the system to become unresponsive to the non-permissive conditions of the kill switch system. This could pose a risk for the escape of the genetically modified bacteria in the environment, which is a widespread societal concern.
While we observed the emergence of these escape mutants with the pKA1 and pKC1 system at high IPTG concentrations (high toxin expression), longer growth inhibitions were observed when the expression of the toxin was reduced. While resulting in some growth of the bacteria, the growth rate was largely reduced. Considering the competitive environment of the gut microbiota and its quick turnover rate [12 ,13], the longer growth inhibition observed at lower toxin expression induction are most likely sufficient to guarantee the removal of the bacteria from the patient’s gut. Once in the environment (for example the wastewater system), where the temperature is likely even lower than 25°C, we expect the bacteria to not proliferate, as we clearly demonstrated that the drastically reduced growth rate at 25°C persists for at least 10 hours. To validate these hypotheses we suggest to firstly measure the retention of E. coli Nissle 1917 with one of the kill switch plasmid in an in vitro continuous fermentations with a complex gut microbiota with and without induction of the kill switch system, and secondly to measure the proliferation of E. coli Nissle 1917 within a complex waste water sample. Moreover, our design already includes another component which improves the evolutionary stability of the kill switch system. This component is a cold-inducible RNA thermosensor which can be coupled to the toxin. This would limit the expression of the toxin at 37°C but not at lower temperature. This would further reduce the selective pressure to mutate the toxin (or any other component which would lead to inactivation of the kill switch) under permissive conditions rendering our kill switch system even more robust.
While we demonstrate that our kill switch system can be effectively used for biocontainment in the environment, due to time constraints we have not performed measurements of the design of our system encoding for the phosphate control. However, this component has already been extensively characterized by the NUS 2017 team (http://parts.igem.org/Part:BBa_K2447000), we are therefore confident that it’s implementation would succeed as planned.
As in practice IPTG and aTc cannot be supplied to induce the toxin and antitoxin, the pTet and pTac promoters need to be replaced by either a constitutive or another type of inducible promoter. The selection of these promoters can be easily done using the abundant promoter characterization in the iGEM Registry or in the literature, and can be guided by our kill switch model. Concretely, one can use our measurements to see for which aTc and IPTG concentration the desired growth inhibition effect is obtained. Then, these inducer concentrations can be matched to the GFP-measurements of the pTet (BBa_R0040) and pTac (BBa_K2572025) promoters. The corresponding GFP output can then be matched to that of one of many promoters with GFP dose-response measurements, such as those in the Anderson promoter catalog (http://parts.igem.org/Promoters/Catalog/Anderson).
We demonstrated that all three constructed kill switch systems are able to inhibit cell growth as desired, and also showed for which level of inducer concentrations of the toxin and antitoxin expression different levels of growth inhibition can be obtained. We also argued that our kill switch system is most likely sufficient to guarantee a high level of biocontainment and its translation to a completely autonomous kill switch system for application in clinical settings to be achievable with minor adaptations.
Best Measurement Achievement
With our extensive characterization of the toxin-antitoxin systems ccdB/ccdA (BBa_P1010,
BBa_K1075032) and miniColicin E2/IM2 (BBa_K1976048, BBa_K1976027) we lay the foundations for an easy and standardized implementation of kill switch solutions based on ccdB/ccdA and miniColicinE2/IM2 for the iGEM community.
References
- [1] P. Bernard and M. Couturier, “Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes,” J. Mol. Biol., 1992.
- [2] F. Collin, S. Karkare, and A. Maxwell, “Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives,” Applied Microbiology and Biotechnology. 2011.
- [3] E. Cascales et al., “Colicin Biology,” Microbiol. Mol. Biol. Rev., 2007.
- [4] X. Jin, W. Kightlinger, Y. C. Kwon, and S. H. Hong, “Rapid production and characterization of antimicrobial colicins using Escherichia coli-based cell-free protein synthesis,” Synth. Biol., 2018.
- [5] B. C. Stanton, A. A. K. Nielsen, A. Tamsir, K. Clancy, T. Peterson, and C. A. Voigt, “Genomic mining of prokaryotic repressors for orthogonal logic gates.,” Nat. Chem. Biol., vol. 10, no. 2, pp. 99–105, 2014.
- [6] S. Sen, D. Apurva, R. Satija, D. Siegal, and R. M. Murray, “Design of a Toolbox of RNA Thermometers,” ACS Synth. Biol., vol. 6, no. 8, pp. 1461–1470, 2017.
- [7] A. Hoynes-O’Connor, K. Hinman, L. Kirchner, and T. S. Moon, “De novodesign of heat-repressible RNA thermosensors in E. Coli,” Nucleic Acids Res., vol. 43, no. 12, pp. 6166–6179, 2015.
- [8] J. Tsang, “Bacterial plasmid addiction systems and their implications for antibiotic drug development,” Postdoc J., 2017.
- [9] M. Olier et al., “Genotoxicity of escherichia coli nissle 1917 strain cannot be dissociated from its probiotic activity,” Gut Microbes, vol. 3, no. 6, pp. 501–509, 2012.
- [10] Openwetware, “E. coli genotypes.” [Online]. Available: https://openwetware.org/wiki/E._coli_genotypes.
- [11] D. Duché, A. Frenkian, V. Prima, and R. Lloubès, “Release of immunity protein requires functional endonuclease colicin import machinery,” J. Bacteriol., 2006.
- [12] M. A. Bauer, K. Kainz, D. Carmona-Gutierrez, and F. Madeo, “Microbial wars: Competition in ecological niches and within the microbiome,” Microbial Cell. 2018.
- [13] W. Tottey et al., “Colonic transit time is a driven force of the gut microbiota composition and metabolism: In vitro evidence,” J. Neurogastroenterol. Motil., 2017.