Team:SUNY Oneonta/Model
SUNY Oneonta iGEM
Modeling
Overview
Yeast RAD27 (FEN-1 in humans) is an endonuclease that plays an important role in the removal of a 5’overhanging substrate in DNA. Known as flap endonuclease, or flappase, this protein serves as a key element in maintaining genomic integrity. It is the central component of the Invader assay, which is used to detect single nucleotide polymorphisms (6). We are using flappase in our detector system to detect the A2 allele of the beta-casein gene. We chose to use the yeast gene for practical reasons; yeast enzymes typically function well over a wide range of temperatures, making them well-suited for field applications. However, much of the literature on the Invader assay does not specify the source of the enzyme or uses the human enzyme. The structure of the human enzyme is known, but the structure of RAD27 is not. Therefore, we employed homology modeling to produce a model of flappase from S. cerevisiase using the algorithms implemented in the SWISS-MODEL server (3,4) to provide evidence that methods used with the human enzyme would be transferable to yeast flappase. We also intend to use this model when planning mutations that may be necessary as we improve detector function, for example, when optimizing interactions with target oligonucleotides or trying to understand and interpret the enzymatic activity of the system (12,17).
Homology Modeling
Structural biology is an area of science that focuses on the molecular structure and mechanisms of biological macromolecules, particularly proteins. Within structural biology, there is an area of bioinformatics that focuses on the generation of software and methods used to predict macromolecular structures, known as homology modeling. Homology modelling tools enable scientists to generate a prediction of the three dimensional structure of a protein when only sequence data is available (9,11). Homology models are based on the idea that if two peptide sequences are highly similar then their structures are also similar. Therefore, the quality of the homology model is dependent upon the percent similarity between the model and the template sequence. If the degree of similarity between the two are high, that means the query protein will yield a meaningful result, and vice versa. We built a homology model of the yeast RAD27 protein using the SWISS-MODEL server (11).
Method
We used the following programs and databases to construct, validate, and visualize our homology models of yeast flappase:
- The UniProt database was used to obtain the amino acid sequence of the yeast flappase (16)
- BLAST was used to compare the yeast sequence to proetins of known structure in order to identify a possible template (5)
- SWISS-MODEL was used to produce the model and analyze its validity (3,4).
- UCSF Chimera was used to produce images of our models and analyze the structures via Ramachandran plot (10,13).
We obtained the sequence for yeast flappase from using the UniProt database. Then we performed a BLAST search to locate similar sequences. We identified two potential template candidates with high similarity to the yeast sequence, the Homo sapiens and Pyrococcus furiosus FEN-1 proteins. The sequence alignments are shown in Figure 1. We decided to produce two homology models, one using the human structure (60% identity) as a template and the other with P. furiosus (40% identity) as a template.

Figure 1a- Sequence alignment of yeast and human flappase

Figure 1b- Sequence alignment of yeast and P. furiosus flappase
Next, we used the SWISS-MODEL server to produce the homology models. We had two potential structures to choose from for the human template structure. We chose to use the structure of human FEN-1 when bound to DNA, so that our model would enable us to envision how the flappase interacts with its target. Template PDB codes are given in Table 1 and the template structures are shown in Figure 2.
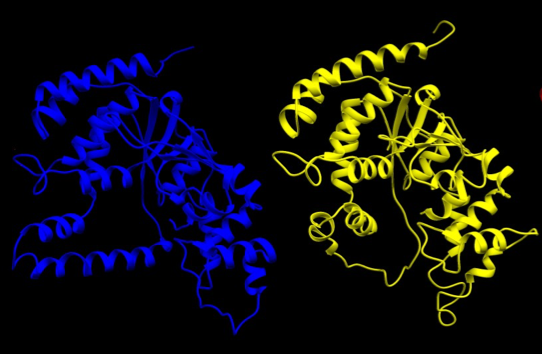
Figure 2- The structures of human FEN-1 in the bound conformation (left, in blue, DNA not shown) and P. furiosus FEN-1 (right, in yellow).
Results
An image of our model superimposed on the human template structure is shown in Figure 3. The quality of the two models was analyzed using standard metrics implemented in SWISS-MODEL. The QMEAN and GMQE scores, shown in Table 1, indicate that the resulting structures are reliable. Ramachandran plots of the models indicate they represent a regular protein structure with most angles falling in the allowed regions (Figure 4 and Table 1). As expected due to the much higher level of sequence identity, the model based on the human structure is of better quality.
Table 1
|
Template Structure |
Sequence Identity |
Quality Data |
Ramachandran Results |
|
P. furiosus (PDB ID: 1B43) |
43.08% |
QMEAN: ( -3.12) GMQE: (0.56) |
Favored: 90.14% Outlier: 2.9% |
|
Homo Sapiens (PDB ID: 5UM9) |
62.31% |
QMEAN: (-2.48) GMQE: (0.61) |
Favored: 92.75% Outlier: 2.42% |
- GMQE (ranges from 0 to 1.0, a higher number is a more reliable model)
- QMEAN (the closer to 0, the higher the model quality; numbers < -4 indicate poor model quality)
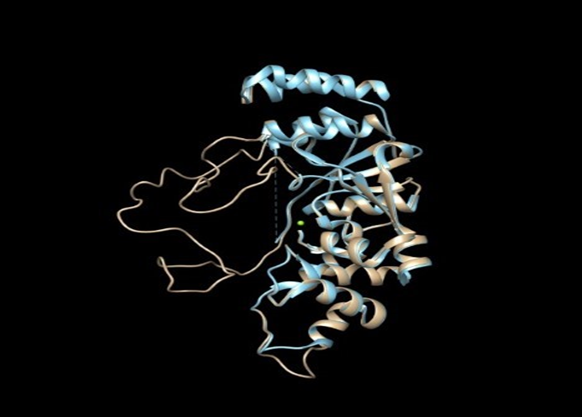
Figure 3- The yeast flappase homology model (tan) superimposed on the human FEN-1 structure (light blue).
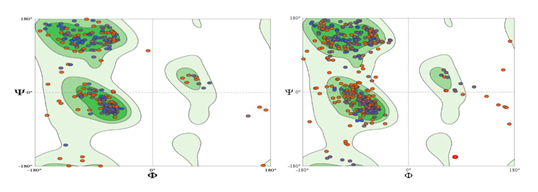
Figure 4- Ramachandran plots of the yeast flappase model based on human FEN-1 (left) and P. furiosus FEN-1 (right)
Future Work
We were able to successfully produce a homology model of the yeast flappase protein. Due to time constraints, we were not able to further validate our model via simulation. In the next phase of our project, we intend to do this. We will also refer to the model when we are considering mutations to optimize the function of flappase in our detector.
References
-
Al Ait, L., Yamak, Z., & Morgenstern, B. (2013). DIALIGN at GOBICS--multiple sequence alignment using various sources of external information. Nucleic acids research, 41(Web Server issue), W3–W7. https://doi.org/10.1093/nar/gkt283
-
Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011 Feb 1;27(3):343-50. doi: 10.1093/bioinformatics/btq662. Epub 2010 Dec 5. PMID: 21134891; PMCID: PMC3031035.
-
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Gallo Cassarino, T., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research, 42(Web Server issue), W252–W258. https://doi.org/10.1093/nar/gku340
-
Bienert, S., Waterhouse, A., de Beer, T. A., Tauriello, G., Studer, G., Bordoli, L., & Schwede, T. (2017). The SWISS-MODEL Repository-new features and functionality. Nucleic acids research, 45(D1), D313–D319. https://doi.org/10.1093/nar/gkw1132
-
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: architecture and applications. BMC bioinformatics, 10, 421. https://doi.org/10.1186/1471-2105-10-421
-
Hosfield, D. J., Mol, C. D., Shen, B., & Tainer, J. A. (1998). Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell, 95(1), 135–146. https://doi.org/10.1016/s0092-8674(00)81789-4
-
Liu, Y., Kao, H. I., & Bambara, R. A. (2004). Flap endonuclease 1: a central component of DNA metabolism. Annual review of biochemistry, 73, 589–615. https://doi.org/10.1146/annurev.biochem.73.012803.092453
-
Lyamichev, V., & Neri, B. (2003). Invader assay for SNP genotyping. Methods in molecular biology (Clifton, N.J.), 212, 229–240. https://doi.org/10.1385/1-59259-327-5:229
-
Martí-Renom, M. A., Stuart, A. C., Fiser, A., Sánchez, R., Melo, F., & Sali, A. (2000). Comparative protein structure modeling of genes and genomes. Annual review of biophysics and biomolecular structure, 29, 291–325. https://doi.org/10.1146/annurev.biophys.29.1.291
-
Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
-
Muhammed, M. T., & Aki-Yalcin, E. (2019). Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chemical biology & drug design, 93(1), 12–20. https://doi.org/10.1111/cbdd.13388
-
Ng, P. C., & Henikoff, S. (2006). Predicting the effects of amino acid substitutions on protein function. Annual review of genomics and human genetics, 7, 61–80. https://doi.org/10.1146/annurev.genom.7.080505.115630
-
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry, 25(13), 1605–1612. https://doi.org/10.1002/jcc.20084
-
Ramachandran, G. N., Ramakrishnan, C., & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. Journal of molecular biology, 7, 95–99. https://doi.org/10.1016/s0022-2836(63)80023-6
-
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F. T., de Beer, T., Rempfer, C., Bordoli, L., Lepore, R., & Schwede, T. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research, 46(W1), W296–W303. https://doi.org/10.1093/nar/gky427
-
UniProt Consortium (2019). UniProt: a worldwide hub of protein knowledge. Nucleic acids research, 47(D1), D506–D515. https://doi.org/10.1093/nar/gky1049
-
Xie, Y., Liu, Y., Argueso, J. L., Henricksen, L. A., Kao, H. I., Bambara, R. A., & Alani, E. (2001). Identification of rad27 mutations that confer differential defects in mutation avoidance, repeat tract instability, and flap cleavage. Molecular and cellular biology, 21(15), 4889–4899. https://doi.org/10.1128/MCB.21.15.4889-4899.2001
-
Xu, H., Shi, R., Han, W., Cheng, J., Xu, X., Cheng, K., Wang, L., Tian, B., Zheng, L., Shen, B., Hua, Y., & Zhao, Y. (2018). Structural basis of 5' flap recognition and protein-protein interactions of human flap endonuclease 1. Nucleic acids research, 46(21), 11315–11325. https://doi.org/10.1093/nar/gky911
Competition Year: 2020 Email: iGEM@oneonta.edu 2020.igem.org