Team:UZurich/Notebook
Notebook
Notes
06/29
Franka
First Day in Lab!
Transformation of LI plasmids (p1-p34) into competent E.coli cells.
Plating Agar medium with corresponding antibiotics (add 400 uL of GENT, SPEC, KANA) to the liquid agar.

06/30
Elio, Nic
Colony Picking and inoculating LB.
Discuss with Kyle the Primer design (Esp3I vs Bpil). Some plates (18,21,22,25,31,32) were too confluent to pick a single colony, thus the bacteria from these colonies were onto new plates and incubated overnight. Colonies from these plates need to be picked tomorrow.
Made TAP Media (chlamy)
Solution #1: TAP salts solution
Solution #2: Phosphate solution
07/01
Nic, Jonas
TAP Media preparation was continued and colonies picked:
1l was generated, whereby 50ml of the media was added to 4 X 200ml Erlenmeyers respectively, 2x 250ml in a glass bottle (1l total volume) with 3.75g of agar added respectively as these will be used for solid media. The remaining 300ml were also poured into a 1l glass bottle.
Miniprep and colony picking: Colonies except for (18,21,22,25,31,32) get miniprepped (ref. the amplification protocol) colonies on plates 18,21,22,25,31,32 get picked
07/02
Elio, Michelle
Miniprep & transformation of FLS2 and CORE
Performed miniprep of plasmids 18,21,22,25,31 and 32 according to protocol.
Transformed DH10-Beta cells with plasmids containing CORE and FLS2 according to protocol onto Spec plates.
Measure DNA content of miniprep by NANODROP
07/03
Michelle, Jonas
Repeat transformation of FLS2 and CORE, primers arrived today, lated our chlamy
Checked for DNA concentration of the sent CORE and FLS2 DNA in the tubes: The concentration was rather low (12 and 20 ng/μL), but it should be enough
Repeat the transformation according to the protocol with the following difference:
Step 2: add 2 μL of plasmid and put on ice for 30 minutes
Also bacteria that weren't plated were stored at 4 degrees
Scrap off colonies of CC1690 and inoculated a total of 3 plates, inoculated liquid colony in Erlenmeyer Flask as well (120 RPM)
07/06
Elio, Michelle
Picked the two colonies of FLS2 and transferred them to LB.
PCR: As an exercise, we attempted to do an PCR of the 34 Plasmids from Germany, not to sequence but just to amplify. We first identified all primers which could bind to a certain plasmid using Snapgene, then see PCR protocol.
07/07
Elio, Michelle
Ordered primers
Miniprep of yesterday picked FLS 2 nanodrop them: 152.0 ng/μL (1) and 185.1 ng/ μL (2)
Check our PCR products via gel electrophoresis: The PCR products from 06-07 were analyzed according to protocol but did not show nice bands, probably due to miscalculated solution quantities. Anyway one could see that there has been some DNA amplification, and the purpose was to get to know the technique.
Yeast media according protocol, make 40% glucose stock solution
07/08
Elio
Sequencing intro with Kyle, order CORE (via IDT).
The following plasmids with DNA content significantly above the 200 ng threshold should be diluted 50:50 with sterile water:
4(489), 5(437), 6(447), 7(432), 8(405), 10(377), 11(406), 12(379), 14(522), 16(305), 18(305), 19(429), 21(340), 22(510), 25(382), 28(336), 31(387)
5 microlitres of plasmid, 5 of water
Labelling of container: PL #d and stored with the regular plasmids. The sequencing primers are at a 100 micromolar concentration, but should be diluted to 5 with sterile water.Reference "primer dilutions" table for the volume (100 in total), those will be stored together with the other primers.

07/10
Jonas, Michelle
Sequencing analysis, Chlamy liquid culture, Chlamy hemocytometer
Sequencing general comments: It seems that there is something systemic going on with the ColE1 Seq F primer. My recommendation would be to resequence two or three of the plasmids with this primer that failed - if we get the same results, we can try to design a new sequencing primer ourselves.
Hemocytometer to count cells:
Make 1:10 dilutions: 1 ml of liquid-grown chlamydomonas for the 100 tube and then make 10-1 - 10-3 dilutions with 900 μl of sterile water and 100 μl of the proceeding dilution
Make sure to pipette up and down so that the cells are mixed well in the tube, before pipetting 100 μl to each of the two middle notches.
Count the cells in the 5 big diagonal squares engraved in the hemacytometer.
If there are cells that touch a border: count eg. only top horizontal and vertical left as 1 and rest boundaries as 0: we counted with the 10-3 dilution: 27 cells and 0 cells.
07/13
Elio, Michelle
Transformation of Kyle's plasmids (EFR, BAK1)
Discuss primers for chlamy with Isabel, order chlamy primers
2 overnight cultures: added 5 μl Spec to each, 25 μl E.coli in each tube with EFR and BAK1 DNA, 10 μl of mixed E.coli + DNA to each of the ON tubes
2 plates with Spec: for colony picking tomorrow, 50 μl of mixed DNA + E.coli per plate
07/14
Michelle, Philip
Miniprep BAK1 and EFR
PCR from miniprep BAK1 and EFR
PCR from FLS2(1), primers arrived today
GEL electrophoresis of the PCR products. We need to do PCR for each receptor 2 times since there are always two forward primers (one with and one without signalpeptide) + 1 reverse primer

07/15
Philip, Jonas
Purify the PCR products from 07/14
The vector where we clone PCR products in FLS2 is cut with Esp3l and inserted into 02 p641 Esp3l, EFR is cut with Bpil and inserted into 01 p641 Bpil, Bak1 is cut with Esp3l and inserted into 02 p641 Esp3l
Did a new PCR for the FLS2 with signaling peptide (number 8) that failed to produce anything according to the nanodrop measurements.
Changed the polymerisation phase to a length of 3:30, as FLS2 is 3.5 kb long, so the Polymerase has enough time.
07/16
Jonas, Nic
Transformation of E. Coli with Vectors p641-Esp3I: FLS2-, p641-Esp3I : BAK1+- p641-BPiI :EFR+-
Resequence strange results
PCR Gel of FLS2 with SP: The band in the second column is just the primers, if we had our receptor in the PCR product, the band would be much higher (around 3000 bp). We should have a look at our primers and make sure that they are suitable and reconsider the PCR settings, e.g. decrease the annealing temperature, this should make the primers bind more, with the risk of the DNA strands rejoining.
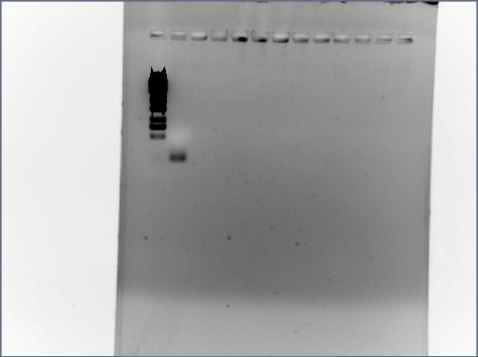
07/17
Nic, Michelle
Colony PCR of vector p641-Esp3I: FLS2-, p641-Esp3I : BAK1+- p641-BPiI :EFR+- (use primers of vector and insert)
Redo p641-Esp3I:FLS2 (1) + SP PCR (check Primers, decrease annealing temp.) NOT SUCCESSFUL
Gel:
top row: 1kb ladder | 100bp ladder | E+(1)| E+(2)| positive control | negative control | F+(1) | F+(2)|
bottom row (COLONY PCR): 1kb ladder | 100bp ladder | F-(1)| F-(2) | B+(1)| B+(2)| B-(1)| B-(2)|E-(1)| E-(2)|
B-(2) and E-(2) are the only ones that show appropriate bands. This means that these colonies can be used for the next step!
The rest should be repeated on the next lab day (Monday 20th july)
07/20
Elio, Michelle
Redo Colony PCR of p641-Esp3I: FLS2- , p641-Esp3I: BAK1+ p641-BPiI:EFR+
Redo PCR of p641-Esp3I:FLS2 (1)+ , and p641-Esp3I:FLS2 (2)+
Created a new program with decreased annealing temp: called PCR X7long50
Diluted the arrived for alpha factor peptide and CORE
DIGLIG of CORE and ALPHA signal peptide
07/21
Michelle, Philip
Miniprep and Diagnostic Restriction Digestio (E+ B+ F-)
Overnight cultures: FLS-, BAK1+, EFR+, miniprep
Sequence EFR+ and BAK1+
Compared gel with theoretical bands from Snapgene simulation. The bands are a little messy, but EFR+ and BAK1+ generally look like they should. FLS2; no idea what happened, the rest proceeds to get sequenced.

07/22
Philip, Jonas
CORE & MFα transformation into DH10beta E. coli
07/23
Jonas, Nic
Overnight culture of EFR- and BAK1-
Colony PCR of Ll FLS2-
RESULTS: First two columns of both rows show the 1kb ladder. The first 8 columns show the colony PCR results of FLS2-. The next column was not loaded. The next 6 show the results for the CORE colony PCR. On the bottom row, the other two CORE colony PCRs can be seen. Again, one column was left empty, to make it easier to analyze the gel. The remaining 8 show the results for the MFα Colony PCR.
CORE: use colonies 1,3,5,7 and 8. ( = pick 2, should be enough for future work)
FLS2-: not good, but it was advised to still continue
MFa: did work but not very nice observable, use 100 ladder next time and run longer = better separation of the bands -> check with in silico
Sequencing Analysis of Bak1+ and EFR+: Alignments didn't cover the full receptor. In the EFR+ there's a mismatch that changes the amino acid. We should probably pick a new colony for the EFR(+). The BAK1+ needs to be resequenced with different primers that also bind to regions within the receptor sequence.

07/24
Elio, Philip
Colony PCR p01:E-, p:02:B-, pouring YPD
Miniprep of p(02, B-) , p(01, E-) was not possible as the overnight culture didn't work
Miniprep FLS2-, MFa, CORE (total 6 as we have 2 cultures/ part)
Pick new colonies for BAK1- and EFR- and do colony PCR: The gel failed completely, only the ladder was visible. Will have to make a liquid overnight culture of all 8 (4 BAK1, 4 EFR) on Monday and do a DRD on Tuesday
07/27
Elio, Michelle
Ordered and design sequencing primers for FLS2 and CORE
Make overnight culture from each two = 4 (2 BAK1-, 2 EFR-) quadrants for DRD on Tuesday
Make more Yeast media for plates
07/28
Michelle, Philip
Miniprep p01:EFR- P02:BAK1-
DRD of ON BAK1- and EFR-
Sequencing (p01:E+,p02:FLS-,p02:CORE, p02:MFa, p02:BAK1-,p01,E-, p02:BAK1+)

07/29
Philip, Jonas
Colony PCR of EFR- and BAK- -> overnight cultures of them
Gentamycin plates
07/30
Jonas, Nic
Replating of 16.7 BAK1- onto 2 new plates
Redo colony PCR for BAK1- and EFR- (either from the ones that were plated on the 29.07 or from the original plate)
EFR-: 16.7 (original) was used (8X), BAK1-: replated colonies from 29.07 were used (2X)
Run Gel of colony PCR BAK1- and EFR- -> SUCCESSFUL
The second column contained BAK1-. Here we see a clear, thick band, meaning that this colony PCR worked successfully! EFR-, although using 8 colonies, only worked once, namely in column nb. 6.
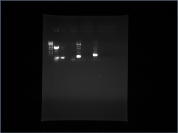
07/31
Elio, Nic
Make miniprep of liquid culture BAK1+/-, MFα and EFR- (preparation of DigLig)
Redo EFR+ colony PCR -> Successful
po2::BAK1- : sequencing looks fine
po2::BAK1+ : sequencing looks fine
po2::MFα : sequencing looks fine
po1::EFR- : looks empty -> Use overnight culture from 30.07 (successful colony PCR) and MiniPrep it
po1::EFR+: looks empty
What happend to po1::EFR+/- : it seems as if the primer dimers were inserted. do 16 colony PCRs from EFR + to search for the correct insert. Out of 16 colony PCRs, one worked namely nb. 9

08/03
Elio, Michelle
Miniprep EFR+, poured YPD plates
Sequence analysis of FLS2- and CORE : CORE fine, FLS2- not so 24 new colonies picked
Check if Kan works as yeast resistance marker: inoculated yeast on one plate with kan and one without -> put in 28 °C incubator overnight
08/04
Elio, Michelle
Sequencing EFR-+, Bak-, FLS-(1), FLS-(2)
FLS2 PCR amplification of both templates (FLS2(1) and FLS2(2))->GEL Purification-> DIGLIG-> then put DigLig in LI p01 in E.coli -> did not work because someone changed the X7long protocol on the machine! We did it again with the correct protocol.
08/05
Elio, Michelle
PCR of FLS2 failed again-> decision with Kyle to order it via IDT
DIGLIG of LII p(10:C) p(10: B+), P(10:B-)
Transformed DH10b to amplify YFP, BB6, BB9
The yeast KAN restistance marker did not work! we will need to use other LII vector than p(11) -> we use dropout medium p(10) with TRP
08/06
Jonas, Nic
Transformation LII p(10:C), p(10:B-), p(10:B+)
Replate YFP, BB6, BB9, restreak of yeast (03.08), restreak chlamy
Seq.Anlys. 04-08:
BAK1 (in p02)(-) : good coverage, looks good
FLS2.1 (in p02)(-): bad coverage, primer dimer insert
FLS2.2 (in p02)(-): bad coverage, primer dimer insert, isn't the end of the world, since we will order FLS2 anyways
EFR (in p01)(-): bad coverage, primer dimer insert
EFR(in p01)(+): bad coverage, primer dimer insert
reamplify from template, gel purification
08/07
Elio, Michelle
PCR Amplification of EFR +/-
Gel product of amplified EFR +/-, with 1kb ladder:-> did not work (Bands are of 500-700 bp.) -> redo PCR Amplification of EFR +/-
Sigma Aldrich: order Yeast Dropout media components
p(10:C), p(10:B+) plates were without any colonies: Since the transformation only worked with p(10:B-) (stored in the fridge), we Repeat DigLig of LII constructs: p(10:C), p(10:B+)
08/10
POWER SHUTDOWN: Overnight cultures BB6, BB9, YFP, BAK1-
08/11
POWER SHUTDOWN: Miniprep of BB6, BB9, YFP, BAK1-
DRD of BAK1-
08/12
Philip, Jonas
Repeat transformation of p10(BAK+) and p10(CORE)
DRD p10(BAK-): inconclusive
Diglig of amplified EFR+ and EFR- into p01 and B+,B-,E- into p10
08/13
Elio, Michelle
DigLig EFR +/-
Transformation: BAK1+/-, CORE in LII and EFR+/- into LI
Overnight cultures of only p(10:B+) since the only one with colonies
08/14
Elio, Nic
Miniprep p(10:B+) & DRD p(10:B+) -> not successful! But plated it anyway to wait for sequencing
DigLig: p(10:CORE) A&B, p(10:BAK1-), p(10:BAK1+)
P(10:B+,B-,C-) and P(01:E-,E+) out of the incubator-> only p(01:E-) successfull
Checkout the dropout: Yeast plates looks fine. there is a little "leakiness" on the ones containing the dropout, however this is okey. No leakiness would be better, however.
08/17
Elio, Michelle
EFR (-) LI colony picking & overnight cultures (use plates preferably from 13.08)
B+ B- LII colony picking and overnight cultures
PCR amplification with the newly arrived primer pr(38) for obtaining Bak1 ectodomain only
Repeat Diglig of LII Bak+,Bak-,CORE
Make new stock dilutions for LII parts
Transformation p(10:B+), p(10:B-), p(10:C) (8 total) (from Fr. 14.08)
Transformation of our todays DIGLIG p(10:B+), p(10:B-), p(10:C) total 6 (from 17-08)
08/18
Michelle, Philip
DigLig new p(10:B+), p(10:B-), p(10:C) -> Transformation of p(10:B+), p(10:B-), p(10:C)
EFR (- ) LI and B(+) B(-) LII each two overnight cultures from 17-08: miniprep, diagnostic restriction (Also do DRD of the p10 plasmid as a control)
Sequence the p10(B-) and p10(B+)
Couldn't pick colonies from 17.8 with p(10:C), p(10:B-), p(10:B+) cause Plates showed no growth
From Bak1 ectodomain PCR from 17.8.20: GEL purification (BAK1 ectodomain is about 850 bp) -> DIGLIG in p02
08/19
Philip, Jonas
DigLig of LI p(01:E-), p(01:E+)
Transformation of Ll DigLig products p(02:B-), p(02:B+), p(02:B*)
Plates from 18.08 were completely empty today, no colonies
Redo the transformation of yesterdays DigLig with more than 1 microlitre of plasmid
08/20
Jonas, Nic
Sequencing analysis: GFP restriction sites are wrong! Seq matches G53 not G12, we have the wrong GFP!
DigLig:The plates with the Bak ectodomain transformed bacteria from 19-08 were completely empty, so the DigLig has to be redone
Transform EFR from 19-08
08/21
Elio, Michelle
DigLig p(10:B+), p(10:B-), p(10:C) and transformation
DigLig and transform(p02: cheA) (p02: cheB), colony PCR EFR +- and eBak
DigLig split mcherry into p02
Check plates for p(02:E+/-) colonies
Colony PCR: p(02:EFR +/-), Colony PCR: p(02: eBak)
1000 Ladder | 100 ladder | E- E- E- E - E - E+ E+ E+ E+ -> here all E should be a band at 3000 bp only the last EFR seems somehow oke: we streaked out the last EFR + on a plate with gent, no EFR - was working so no plate of that.
1000 Ladder | 100 ladder | eB* eB* eB+ eB+ eB-eB- positive control | negative control -> here all bands should be at 1000 bp what they are jupii! we streaked out the eBak s on plates with gent p(P02: eB *) (p02: eB-) (p02: eB+)

08/24
Elio, Jonas
Restreak our yeast on a new YPD plate as the colonies are very big now
Redo colony PCR from p01:EFR- > gel it: gel looks bad, made a new Diglig
p02:eBak pick colonies and transformed it
Repeat LI DigLig of p(01:E-) and transformation
make negative control plate for p02, make overnight cultures of p(01:E+)
EFR showed no amplification: probably wise to drop this plate and wait for todays DigLig and look if that worked
mCherry shows correct amplification, with mCherry A at the bottom left and mCherry B at the bottom right

08/25
Jonas, Nic
cPCR p(01:E-), yeast transformation p(10:B+/-, C-), DRD LII p(10:B+/-, C-), Sequencing p(10:B+/-, C-), p(02:mCherry), p(eB-/+/*)
DRD of LII constructs and eBAK
Sequencing of eBAK, LII constructs and p(02:E+), mCherry
colony PCR of p(01:E-): p(01:E+) = Positive results, p(01:E-) = negative results
Redo DigLig of p(02:E+)
08/26
Elio, Jonas
DRD p(10:B+/-, C-) -> All BAK1 columns look splendid! CORE, however, is just weird. We repeat the DRD for CORE
Dilution series for our yeast strain: Do dilution series on selective and non-selective media. Start at OD 0.2 and do a 10fold dilution 5 times = 1, 1:10, 1:100, 1:1000, 1:10000, 1:100000. This should give us a good feel for how much growth ("baseline") we can expect
Diglig LII E-, FLS2 and eBAK for chlamy and transformation
08/27
Jonas, Nic
cPCR p(01:E-), Yeast Transformation (YT) p(10:B-/+, CORE)
Sequencing Analysis:
p(10:B-): looks good
p(10:CORE): no reads at all(?), p(10:B+): strange results, looks like p(10:B-) instead of p(10:B+) as native signal peptide is missing, error could have arisen while setting up the sequencing reaction or the DigLig
p(02:mCherry): 1-159 looks good, 160-235 looks good
p(02:eB+/-/*): p(02:eB+/*) both look good, no reads for p(02:eB-)
Yeast transformation p(10:B-)
cPCR p(01:E-) use plate from 24.08-> 1 successful
DigLig: p(02:FLS2), eBAK1- chlamy, p(10:mCherry//eBAK1+/-/*) (dummies for - and *, MFα for +)
Overnight cultures of eBAK1-C and FLS2 (from 26.08 plates)
Successful cPCR p(01:E-)
DRD of p(10:CORE-): The "only Scal" and the negative control (no enzymes) look the same. this means that ScaI does not cut, or at least with a very low efficiency! This then results in wierd patterns on the gel. Further, since the sequencing of this plasmid had no reads, we can assume that something with our assembly went wrong. For example in the 2nd column we have 4 bands, one of. course being the uncut, supercoiled plasmid. This means that a multiple of the same insert has been inserted during the assembly.
-> Redo DigLig p(10:CORE)
08/28
Philip, Michelle
DIGLIG and transformation of p(10:Core)
Transformation of p(02:FLS2-), p(02:eBAK1), p(10:mCherry//eBAK1), p(10:B+)
DRD from all the miniprepped overnight cultures-> not good
Asked Isa about the dropout plates: The yeast dropout seemed to work: there was only little growth on the TRP- plate
08/31
Elio, Nic
Plates: all p10(eB-)::splitMcherry look fine p10(eB° mCheA has few colonies though so will not use these)
p2(FLS2) and p2(eBAK) for chlamy are bad, Just smears and no colonies. Kyle said that either the ESP3l enzyme has died or the Gent plate are bad (Gent could have been added too early) yeast with p10(B-): only 2 colonies have grown on 5 plates and one of them looks like Ecoli. The colonies also aren't red, but they should be. Probably means that the transformation failed
Diglig FLS2 and eBAK for chlamy
Overnight cultures from successful p(01:E-) cPCR from 27.05-> miniprep tuesday + sequencing -> thursday Lll
1 Overnight culture of each eBAK+/-/°::McheA/B
4 Overnight cultures of p10( CORE)
4 Overnight cultures of p10(B+)
2 Overnight cultures of p01(E-)
09/01
Elio, Philip
Miniprep everything
DRD everything
split mcherry for chlamy and MFa with Kozak sequence arrived
Diglig split mcherry for chlamy and MFa with Kozak sequence

09/02
Philip, Jonas
Sequencing: Sequencing eBAK +/-/* mCherry, CORE and BAK1+ from yesterdays DRD
Take restreaked FLS2- & chlamy eBAK plates from incubator: There was way too much bacteria on the restreak, have to do the restreak again: FLS2, eBAK, mCherry and MFa
Miniprep EFR-
Check negative plate to see if Gent AB works -> Negative was good, no colonies
DRD p(02:E-): bad, no insert
IDT order for all chlamy stuff arrived -> resuspend and nanodrop
09/03
Jonas
DigLig of EFR-Chlamy, CORE-Chlamy, repeat DigLig for E-, Mfa, mCheA-Chlamy, mCheB-Chlamy, eBAK1R and FLS2
Transformation of DigLig products
Our Yeast plates didn't show any yeast again: Kyle will talk with Laura about her yeast transformation protocol and see how it compares to ours.
Colony PCR: Only p02(Mfa-Kozak), p02(mCherA-Chlamy) and p02(mCherB-Chlamy) showed pickable colonies. So a colony pcr was done for those-> mCherB and the Mfa look fine. 2 ONs were made of each.

09/04
Nic, Elio
Repeat Diglig for eEFR, eFLS, eCORE, eBAK f. chlamy, CheA f. chlamy
Yeast transformation with CORE, BAK1+, BAK1-mCherry (p10(CORE:YFP A1/B1 and B2) p10(eBAK+::mcheB), p10(eBAK°::mcheA) p10(BAK+::YFP 2 and 3))
Check transformations from 03.09 (p(01:E-), eBAK1-Chlamy, eFLS2, eCORE-Chlamy, eEFR-Chlamy, mCherA, mCherB, MFa)-> Colony PCR EFR-
Sequencing: Only usable p10(BAK::splitmCherry) are p10(BAK°::MchA) and p10(BAK+::MchB), both p10(BAK+::YFP) are good.
p10(CORE::YFP) very interesting. CORE B1/B2/A1 are pretty good except in the YFP, there is apparently an insertion of an Adenine in all 3 reads at the same position. But the L1 plasmid of YFP sequencing doesn't show this insertion, so this is probably just a sequencing error.
09/07
Elio, Michelle
Made a new Diglig for CORE into p02, rest picked colonies and made overnight cultures
Yeast overnight cultures
PCR of Nanoluc with Primers which include the FLAG tag
Cylce purification of PCR of Nanolug with FLAG tag
This GEL looks really nice! all the bands are there to be cut out and purified according the protocol:after the DNA extraction there was no DNA in the samples, something must have went wrong -> Kyle said we should use more DNA (since te PJP32 was also very weak in bands)
PCR amplification to include the FLAG tag: into both the Registry template of Nanoluc and our codon optimized chlamy version was done according PCR protocol as well. To purify this we needed to take a Kit provided by Kyle from the lab called Omega cycle pure Kit. Ee run a GEL anyway to see if it worked, then we did a DRD like Restriction but with the Enzymes that cut only at the ends to make the correct ends for the assembly and we also cut the plasmid PJP32 with the same enzymes (BamHi ad XhoI)
Digest and ligate separate into pJP32
Diglig of CORE::mcheA and McheB
New Diglig of eCORE into p02 -> p(02: C)
Colony PCR from plates 04-09: eBak, eFLS2, eEFR, cheA, PCR of Nanoluc to include FLAG, and cycle purification and GEL purification of chlamy and Reg: the Baks look good, the mcherryA strange-> need to do it again.
FLS2 and EFR size of the fragments 2100 and 2700 look well but sequence anyway so. the nanolucFLAGregistry is fine but the nanolucFLAG chlamy not good but correct size so proceed anyway! ON (Gent Resistance since LII) from all the good stuff: all B1, B4, no mcherry!
09/08
Elio, Michelle
Sequencing of MfaK, mcherryB1, p02, eBak, eFLS2, eEFR for Chlamy miniprep from 04-09, p02 plasmid, p02eBak1, p022Bak4, eEFR (1, 2, 3, 4), eFLS2 (1, 4)
Plate the new eCORE from yesterday
PCR of Nanoluc to insert FLAG Tag in both registry-luc and our codon optimized version chlamy-luc-> did not work again!
another round: PCR for insertion of FLAG into chlamy (need the primer mix 15 (pr 52 and 51) did not work again now for the third time!
Cycle purification after the pcr (of Nanoluc to insert Flag tag) to clean up the pcr Product: see protocol cycle pure kit.
NOTE: The PJP32 colony picked ON culture did not work since our plate is dried out and according to Kyle all colonies are ded so we need to do a new ON culture: we can do this directly with the original plasmid transformed in bacteria, since we know its corect with the original PJP32.
Microscope the yeast ON cultures: Bak+, Bak- both show nice expressions! CORE shows no expression
09/09
Philip, Jonas
Transformation of PJP32 vector into bacteria and streaked onto Carb plates
Colony PCR of p(02:eCORE): The gel was completely empty.
PCR of NLuc-Chlamy: ng/ul yield is very low (5.5 and 4). -> This low a concentration is useless for the next step, something went wrong with the PCR again. We discussed it with Kyle-> change the PCR protocol, longer 1 minute seperation step is so that the GC-rich nanoLuc comes apart correctly, 30 seconds in the normal protocol might be to short.
Digestion: Kyle suggested we should digest all of our PCR amplifications: also have to digest the pJP32. We used the one from Joao and this didn't work very well so maybe try it with the pJP32 that we minipreped. digest about 1 ug of pJP32.
Gel purification : when we did it yesterday, we didn't change the running buffer. Also, when we nanodroped our gel purified stuff, we got a high peak at 230 nm, which indicates contamination, probably because we didn't cut away enough excess gel.
09/10
09/11
09/14
Elio
Sequencing results: G51 and G52 are the bad GFP with the wrong overhang sequences. G53 is the good YFP, keep it. eFLS: pretty strange: eFLS 1 and eFLS4 have two deletions which would cause frameshift mutations, which means they would be useless. Make Colony-PCR of eFLS2 again. eEFR 1-4: all but number 4 seem pretty good. eEFR 4 has a SNP G -> A, but this is a silent mutation, which chlamy doesn't like, so don't use this one.
eBAK for chlamy 1 and 2. eBAK f. chl. 1 has a large deletion. eBAK f. chl. 2 has a good reverse primer sequence, but the forward read is wrong, there seems to be contamination as you can see multiple peaks overlap. But it still seems that eBAK f. chl. 2 is good, so use this one. p02(McheB for chlamy) is perfect, empty p02 is good
Make Colony-PCR of eFLS2 again and ON (bands 2, 5, 6 and 8 look fine)
Diglig: p11(eEFR::McheA (not for chlamy), p10(eEFR::YFP), p11(CORE::mcheA(not for chlamy)) p10(CORE::YFP) (as the Yeast colonies transfected with this plasmid didn't glow) P2 (McheA for chlamy) p2(CORE) (from the 3 fragments). We need the p11 plasmid be

09/15
Elio, Nic
Miniprep eFLS2 (4 overnight cultures)
LBit reg, LBit yeast, LBit chlamy, sBit reg, sBit yeast, sBit chlamy, FRB and FKBP12 arrived, resuspend and diglig into P2
Amplification of BAK-::YFPG for in fusion cloning
09/16
Elio
CPCR of p02(mcheA) all the split lucs and the FRB, FKBP12
In Fusion cloning of BAK1::YFP into pJP32 and Diglig of Registry nanoluc into pJP32
Picked first largebit registry, first largebit yeast and second largebit chlamy and second McherryA for chlamy. Because the ladders were quite bad, we couldn't determine the size of the smallbits, so do again. FRBs and FKBP12 very weird, as for example the first and secon FRB have different size, very weird, repeat tomorrow.

09/17
Elio
Miniprep All Lbits, CheA for chlamy, P11(CORE::MchA), P11(EFR::MchA), p10(CORE:YFP), p10(eEFR:YFP) Lbit yeast and registry didn't grow for some reason, so pick same colony from yesterday again
Colony PCR for Lbit R, all Sbits, p02(eCORE),FRB and FKBP12
Sequence the 4 FLS2
Make overnight culture of WT and p10(BAK::MchB)
Amplify Nanoluc for chlamy with Flagtag primers
Filter sterilize G418
From left to right: 1000 bp ladder, 2x p02(eCORE), 2x Smallbit chlamy, 2x smallbit registry, 2x smallbit yeast, 2x FRB, 2xFKBP12, 2xLargebit registry, 100bp ladder. I made overnight cultures of the SB registry, SB yeast and FRB. eCORE was again negative, suggesting that something is wrong with the IDT ordering. I would suggest dropping it, which isn't too dramatic, as it is only for chlamy.

09/18
Elio
Only did 100 ml instead of 300 ml for the 3 hour growing step as we only need to do 4 transformations for each strain, and 100 ml is enough for 5 transformation. Incubated the p10(eBAK for yeast::mcheB) for 4 hours instead of 3, as it grows slower than the WT in YPD the rest of the protocol remains the same. Just discovered that ammonium sulfate inhibits Geneticin selection, and, of course, YNB contains ammonium sulfate, so could only do transformation with the p10 plasmids.
Now that's a great DRD!. Everything is as it should be! From left to right: 1kb ladder, 2xp10(eEFR::YFP), 2xp11(eEFR::MchA)

09/21
Elio
Make colony PCR of Nluc registry in pJP32
Make colony PCR of SBit for chlamy and LBit for yeast.
Make digligs of p10(eBAK1::YFP for yeast). Make one Digilig withe the new BB9, P24, p34 and YFP and one diglig with the BB9, p24 and p34 Mastermix (use 3 ul of that) make sure to only use eBAK for yeast and not the one for chlamy
Transformation of DigLig products
Check if yeast colonies have grown->Yeast colonies have grown
Re-amplify Nanoluc for chlamy w. flag tag with the primer NLuc_R2-F and NLuc_R2-R, Use anealing temperature of 53°
Make Overnight culture of p10(eBAK::MCheB) in Trp- medium
09/22
Elio, Nic, Michelle
Sequence: Mcherry A for chlamy, eEFR (1)
Do first step of yeast transformation: inoculate 100 ml of -trp with the ON culture
Finish -Trp plates with geneticin (not gentamicin)
Transform yeast: Transformed 2 p11(eEFR::MChA) and p11(CORE::MChA) each into yeast containing p10(BAK::MchB) and plated it onto the freshly prepared -TRP plates + G418
Re Amplify Nanoluc w. FLAG for chlamy using Phusion polymerase
In Fusion cloning: the protocol recommends 100 ng of vector and 50 ng of insert, so added 5 ul of the cut, gel purified pJP32 and 1.5 ul of the amplified BAK::YFP
09/23
Elio
PCR amplify Nluc registry -> did not work!
Clean up Nluc registry and Nluc chlamy
Prepare YPD
Investigate p10(eEFR::YFP) and p10(CORE:YFP) with epifluorescence
09/24
Elio
Make diglig of eEFR::mchb for chlamy, eBAK1::mchA for chlamy. eEFR::SBIT with full length receptor eEFR::SBIT with truncated receptor, eBAK::LBIT with full length receptor, EBAK::LBIT with truncated receptor, CORE::SBIT with full length receptor, CORE::SBIT with truncated receptor
Make dilutions of all the plasmids needed for transformation
ON untransformed yeast in YPD, p10 transformed yeast in -.TRP +geneticin to test effectivity
09/25
Elio
Diglig of yesterdays DNA mixture
Transform yeast with p10(eBAK for yeast:YFP)
PCR of Nanoluc registry to include Flag tag and and reamplification of Nanoluc for chlamy with flagtag with iGEM_Nluc_R2- F and iGEM_Nluc_R2- R -> The DNA content measured via nanodrop for the reamplified Nanoluc was 10 ng/ul, while the one for the registry nanoluc was 33 ng/ul. Only kept the latter as the former is not enough for successfull cloning.
09/28
Elio
Check the yeast plates of p10(eBAK::YFP)-> looks good
Make colony PCR of the p10 and p11 colonies-> ON
Prepare liquid -TRP medium without Ammonium sulfate YNB Inoculate 2x30 ml of this freshly prepared medium with the co-transformed yeast (one for eEFR, one for CORE)
Re-Amplify Nanoluc for chlamy with FLAG
Transform yeast with the purified plasmids


09/29
Nic, Michelle, Elio
Autoclave some flasks for ON and transformation of Yeast
DigLigs of p10(eBAK::mCheA)-Chlamy, p11(CORE::SBit)Full, p11(CORE::SBit)Trun
Miniprep the ONCs from yesterday-> Sequence
Purify the nanoluc pcr from yesterday
Yeast ONs: look in Tecan for fluorescene
Transform yeast with the purified plasmids
09/30
Elio
Clean up PCR of Nluc for chlamy from yesterday ->phusion with the EZNA cycle pure kit and got DNA concentration of around 35 ng/ul for both.-> Unfortunately, Nanoluc for chlamy appears as a smear, which makes it unusable. Nluc registry and pJP32 look fine.
Clone nanolucs into pJP32
Begin the western blot for yeast with p10(BAK::MchB)+p11(CORE/EFR::MchA)-> Also prepared 2 SDS-PAGE gel with 15 wells according to the protocol
10/01
Elio
Repeat PCR of Nluc for chlamy with GC buffer
Overnight culture of p10(eBAK::LBit) full 1 and 2, p10(eBAK::LBit) trunc 2 in -Trp
Run SDS gel and transfer it onto membrane
Make overnight culture of pJP32
DigLig: p10(FRB:Sbit) reg and yeast, p8(FKBP12:LBIT), p10(eBAK::YFP) for chlamy
Restreaked p10(eBak::mchB)
As the PCR of Nanoluc for chlamy proved difficult, I decided that I do the digligs for the Split nanoluc codon optimisation experiments. I also made a diglig for p10(eBAK::YFP) for chlamy so that we might make a localisation experiment in chlamy with eBAK
10/02
Elio
Started the transformation of Bak1::Lbit with full length ADH1 promotor yeast with eEFR::Sbit, but was terminated because of western blot result
Treating of Western blot membrane with αFLAG and αHA
PCR of Nanolucs, terminated because it was realised that there aren't any BPs upstreams of the recognition


10/05
Jonas
cPCR of: (SBit:FRB)reg, (LBit:FKBP)reg, (SBit:FRB)yeast, (LBit:FKBP)yeast, (eBAK:mCheA)chlamy, (eBAK:YFP)chlamy
(SBit:FRB)reg -> all fine, pick 1 and 2
(LBit:FKBP)reg -> all fine , pick 1 and 2
(Sbit:FRB)yeast -> all fine, pick 1 and 2
(LBit:FKBP)yeast -> all fine, pick 1 and 2
(eBAK:mCheA)chlamy -> 3 and 4 (maybe 2) are good, pick 3 and 4
(eBAK:YFP)chlamy -> all are good, pick 1 and 2
(CORE:sBit)full -> all have light bands, will pick 1 and 4

10/06
Elio
Miniprep overnight cultures from yesterday -> sequencing
10/07
Elio
Western blot of eBAK::YFP, BAK::YFP, eBAK::Lbit, eBAK::mchA, eBAK::mchB, (eEFR+eBAK)
Cut pJP32 for inFusion cloning

10/08
Elio, Philip
Yeast transformation: p10(eBAK::Lbit) and p10(eBAK::mchA)
Colony PCR of eEFR::YFP in pJP32
Make Carbenicillin LB plates
Confocal imaging: it seems that the whole YFP expression that we saw is just autofluorescence. Since YFP is PH sensitive and it looks like it's in the vakuole we have a problem!
10/09
Elio, Philip
Yeast transformation
colony PCR of eEFR::YFP in pJP32
PCR of eBAK::YFP and eBAK::mchA for in Fusion cloning
10/12
Jonas
Transform bacteria with pJP32
A lot of overnight cultures
10/13
Elio, Philip
Digestion of pJP32 for in-Fusion cloning in chlamy, something went wrong
Dimerization Assay: Compared fluorescent output of Registry split nanoluc and our codon-optimized version of split-nanoluc.
10/14
Elio, Philip
Restriction Digestion of pJP32
Gel purification of the digestion product , cut out the band at 4900 bp -> In-Fusion cloning
Split-NanoLuc measurement: Results showed higher luminescence for the registry split-NanoLuc than for the codon optimized one
Plate reader fluorescence assay of 5 yeast strains transformed with receptors fused to YFP: BAK1+, BAK1-, ectodomain BAK1, ectodomain EFR, CORE. All transformed strains show higher fluorescence than the negative control!

10/15
Elio, Philip
Chlamydomonas NanoLuc colony PCR
Western blots of YFP constructs
10/16
Elio, Philip
Dimerization assay of yeast strains transformed with split NanoLuc constructs. There seems to be some increase in chemoluminescence after addition of the appriproate elicitor! The split NanoLuc system might be working!
Confocal miscroscope imaging of the YFP constructs: Membrane localization for EFR and BAK1!


10/19
Elio, Jonas
Dimerization assays with EFR & eBAK1 fused to split NanoBit and CORE & eBAK1 fused to split NanoBit
10/20
Elio
Dimerization assay of EFR mcherry and EFR NanoBiT & NanoBiT codon opt.
10/22
Elio
Inoculate -trp medium wit BAK+::YFP, BAK-::YFP,eBAK::YFP,CORE::YFP, eEFR::YFP for imaging tomorrow
FACS measurements


10/23
Elio
Confocal imaging of the cultures from yesterday
Centrifuge all tubes at 2000 g for 5 min, remove supernatant, resuspend in 10ml TE buffer. take 1ml into eppendorf, centrifuge at max speed for 10 seconds. take out 950 ml, then add 1 ul of 2.5 mM FM4-64. incubate on ICE for 1 min, then add 1ml TE, resuspend and the centrifuge. Repeat this wash a second time, then resuspend in 50 ul TE. Keep on Ice until microscopy


