Team:Calgary/Cellulase Engineering
 Overview
Overview
Understanding the Problem
Identifying Cellulases
Part Design
Experimental Design
Preliminary Cellulase Testing
Future Directions
References
OVERVIEW

To allow our target communities to easily grow our yeast strain, we are integrating cellulase genes into the Yarrowia lipolytica genome. This will allow our yeast to be able to extract glucose from readily-available cellulose material to use as an energy source for growth and beta-carotene production.
Our initial goal is to create a proof of concept in the lab. To do this, we followed the engineering design cycle to build our project:
- Understand the Problem
- Research and Ideate Solutions: Identifying cellulases
- Design Solutions: Part design
- Design Solutions: Experimental design
- Create and Test: Preliminary Cellulase Testing
- Evaluate
It is important to note that there will be circular action in this process. Even currently, the results of our Preliminary Cellulase Testing will inform our experiments as we move into genetic engineering. Over the next year, we will be working towards a lab proof of concept for our cellulase integration system, however this engineering design cycle will repeat once we have accomplished this and look towards community implementation. We highlight our plans for next year and beyond iGEM in the Future Directions section.
UNDERSTANDING THE PROBLEM
A lack of sustainable solutions
Nutritional yeasts are well-established as producers of essential vitamins for human consumption. This ranges from pharmaceutical production for supplements, to the consumption of nutritional yeast to treat vitamin deficiencies in WWII. However, to design a yeast that could be cultivated in households and community centres in developing regions introduces a whole new set of challenges. No longer can growth rely on optimized conditions and expensive media. For our idea to be successful, the yeast had to be able to fend for themselves using only the most inexpensive and readily accessible food sources. Our solution was cellulases. Cellulose makes up the cell walls of all plants, and is the most abundant polymer on earth (Bringham 2018). By engineering our cells with a suite of cellulases, we allow the cell to access crude, universally available cellulose as their sole sole fuel source for growth and beta-carotene synthesis.
IDENTIFYING CELLULASES
Why cellulases?
Upon speaking with Dr. Charles Mather, we learned that individuals in our target regions frequently repurpose agricultural waste. From this discussion we were inspired to engineer Yarrowia lipolytica to produce cellulase, allowing VAD communities to sustainably and inexpensively cultivate our yeast using locally available plant matter, such as rice husks and straw. An additional benefit is that cellulase integration provides an environmentally-friendly use for agricultural waste, which is otherwise burned at the end of the harvest season, contributing to greenhouse gas emissions.
The cultivation process using agricultural waste will be performed inside of bioreactors that are based on our design. To facilitate breakdown of agricultural waste inside the bioreactor, it will first be boiled or cut into smaller pieces. Our cellulases will then work to break down cellulose to glucose.
Picking our cellulases
There are three classes of cellulase enzymes necessary for glucose mobilization from cellulose: first, endoglucanases cleave internal beta-bonds at random sites. Next, cellobiohydrolases catalyze the release of cellobiose disaccharides from the reducing and non-reducing ends of the cellulose hydrolysates. Finally, beta-glucosidases break the cellobiose into two glucose molecules (figure 1). We picked cellulases from well-characterized filamentous fungi, which have been used in a number of industrial applications in the past. Furthermore, previous studies integrating cellulases into Y. lipolytica informed our decision to include NcCBHI (BBa_K3629014) in our design and use the promoters and signal peptides that were attached to our cellulase genes (Wei et al 2018).
Figure 1. Degradation of cellulose to glucose using endoglucanases, then cellobiohydralases, and finally betaglucosidases.
PART DESIGN
Optimizing our cellulases
Unfortunately, the most characterized cellulases from filamentous fungi have optimal activity at pHs and temperatures outside the optimal range of growth for Y. lipolytica. Therefore, we employed protein modelling and subsequent protein engineering to modify certain amino acids in these enzymes to make them more stable and functional in Y. lipolytica . Endoglucanase I and cellobiohydrolase I are two essential enzymes for efficient cellulose degradation and are highly produced in T. reesei (Wang et al 2018 and Larry et al 2018). Thus, it is essential these two enzymes in particular are optimized for function in Y. lipolytica.
We used homology modelling and molecular dynamics simulations to create a chimeric Penicillium funiculosum CBHI and a chimeric Trichoderma reesei EGI that have optimal temperature and pH 1 unit above the wild-type enzymes. This presents the first engineered cellulases for Y. lipolytica. Upon further lab access, we hope to add essential experimental characterization to these parts.


Figure 2. Homology model of Modified Trichoderma reesei EGI (BBa_K3629008) on the left and Modified Penicillium funiculosum CBHI (BBa_K3629005) on the right.
To learn more about the modelling that was done for each these enzymes click on the icons above or check out our modelling page . The parts from our part collection that include these two enzymes are:
- BBa_K3629005: Coding sequence of Modified PfCBHI with 6x HIS tag
- BBa_K3629008: Coding sequence of Modified TrEGI with FLAG tag
- BBa_K3629013: Expression construct for modified PfCBHI with 6x HIS tag and gibson homology sequences
- BBa_K 3629016: Expression construct for modified TrEGI with FLAG tag and gibson homology sequences
These are also our featured parts documented on our engineering success page.
Part Components
In addition to submitting individual cellulase genes optimized for Y. lipolytica to the registry, we have also created and submitted fully functional expression constructs for these genes:
- BBa_K3629012: T. reesei CBHII expression construct with gibson homology sequences and FLAG tag
- BBa_K3629013: Modified P. funiculosum CBHI expression construct with gibson homology sequences and 6X His tag
- BBa_K3629014: N. crassa CBHI expression construct with gibson homology sequences and Myc tag
- BBa_K3629016: Modified T. reesei EGI expression construct with gibson homology sequences and 6X His tag
- BBa_K3629017: T. reesei EGII expression construct with gibson homology sequences and FLAG tag
- BBa_K3629018: N. patriciarum BGS expression construct with 6X His tag
Each expression construct has the general structure and features seen in figure 3.

Figure 3. General structure and features of cellulase expression constructs submitted to the registry.
PART FEATURES
- The Translation Elongation Factor 1 (TEF1) promoter is a well-known strong constitutive promoter in Y. lipolytica. Past studies expressing cellulases in Y. lipolytica have used this promoter, but have also suggested using the intronic TEF promoter (TEFin) for stronger expression (Wei et al 2014). Therefore, we included the TEFin promoter in most of our cellulase expression constructs, unless previous literature on specific genes (like EGII) suggested otherwise (Wei et al 2014).
- The Lip2 signal peptide is a well-characterized and commonly used secretion tag (Celińska et al 2018); therefore we decided to attach this signal peptide to most of our constructs with the exception of TrEGII (BBa_K3629017), which had the XRP2 signal peptide as per (Wei et al 2014)
- The affinity tag will not be used for purification, but instead for ELISA analysis. Antibodies specific to our proteins would be expensive to purchase, therefore either a 6x HIS tag, FLAG tag, or Myc tag is attached to the N-terminus of each protein for antibody binding.
- The space with the thrombin cleavage site is present to separate the affinity tag from the coding sequence, but also to remove the tag after translation if it is interfering with the protein's function.
- The coding sequences of all the cellulases were codon-optimized and had their endogenous single peptides removed so it would not interfere with the Y. lipolytica signal peptide.
- The XRP2 terminator was used due to its relatively short sequence.
- Gibson homology sequences were designed so these constructs could assembled in a destination vector (BBa_K3629015), but also so the constructs could be assembled to each other (figure 4).
Learn more about our contributions to the original TEF1 promoter (BBa_K2117000) on our characterization page.
To minimize metabolic burden on the yeast, we will engineer three different strains of Y. lipolytica where each will secrete one of the three cellulase enzyme classes. By co-culturing these strains together, they will be able to synergistically utilize the cellulose present. This system of co-culturing strains secreting one class has been successful in the past with engineered Y. lipolytica strains (Guo et al 2017). Since Y. lipolytica is better equipped to integrate linear pieces of DNA through non-homologous end joining rather than homologous recombination or plasmid maintenance (Niehus X et al 2018), the following gene cassettes will be assembled (using our expression constructs listed above), linearized, and transformed into each strain. The gene cassettes going into each strain will be composed of:
- Y. lipolytica Strain 1: Engineered EGI + TrEGII
- Y. lipolytica Strain 2: Engineered CBHI and/or NcCBHI + CBHII
- Y. lipolytica Strain 3: NpBGS
Using Gibson Assembly
All of our parts, which you can read more about on our part collection page have been designed for modular Gibson Assembly. In other words, each expression construct has multiple unique Gibson homology sequences to be assembled with the other expression constructs in different ways. A specific restriction site was placed at the ends of each homology sequence so that a digestion could be done to expose the ends to T5 exonuclease activity. The restriction sites were:
- XhoI
- NcoI
- BamHI
- BbsI
- SalI
- SmaI
- EcoRV
- HpaI
Check out the registry pages for our expression constructs to see how these restriction enzymes are used.
Figure 4 shows an animation of how our expression constructs will be assembled together and with a nourseothricin selection marker to form the three cellulase gene cassettes mentioned previously. However, this animation presents just one way the expression constructs can be assembled together. Check out our appendix to see all of the different gene cassettes that can be made using our expression constructs.
Figure 4. Gibson Assembly of six cellulase genes into three separate gene cassettes using Nourseothricin (BBa_K3619015) resistance as the destination vector. The different coloured rectangles between the coding sequences represent the different Gibson homology sequences. Initial digestion is done by one of XhoI, NcoI, BamHI, BbsI, SalI, or SmaI. Each of the three gene cassette contains one class of cellulase enzymes. The assembled cassettes are then linearized and transformed into Y. lipolytica creating three different strains.
EXPERIMENTAL DESIGN
Thoughtful design of experiments
Below are detailed descriptions of the planned steps/experiments we will conduct to integrate the cellulase enzymes into the Y. lipolytica< genome. Our first experiments will be carried out in Escherichia coli DH5α for cloning and Gibson Assembly, after which the gene cassettes will be introduced to Y. lipolytica. The experimental workflow will follow the depiction in figure 5.

Figure 5. Experimental workflow of cellulase integration into Yarrowia lipolytica
We have 6 different cellulase enzymes, fortunately, co-culturing of strains each containing ONE class of cellulase (i.e CBH, EG, or BGS) has been done to some success in the past (REF) and will be a necessary proof of concept for us.
Upon consultation from experts during our faculty talk particularly from Dr. Raymond Turner and PhD student Trevor Randall, we decided Gibson Assembly would work best to combine multiple cellulase genes into one cassette for transformation into Y. lipolytica.
- Strain 1: EG cassette= Modified TrEGI + TrEGII + Nourseothricin resistance
- Strain 2: CBH cassette= Modified PfCBHI + TrCBHII + Nourseothricin resistance
- Strain 3: BGS cassette= NpBGS+ Nourseothricin resistance
These gene cassettes will be assembled as per the following table:
Table 1. Table 1. Each part listed in the first column must be individually digested by the corresponding enzyme(s) listed in the second column (i.e BBa_K3629016 is digested with BbsI and SalI). The digested parts can then be placed in the same reaction tube with the Gibson reagents added for the plasmid in column three to form. These plasmids are classified as plasmids made in a single Gibson reaction as the initial digested parts are put in the same reaction tube.
|
Parts to add (DNA Part names) |
Digest parts with1 |
Resulting plasmid (composition) |
|
Strain 1
|
1. BbsI and SalI 2. SalI and SmaI 3. SmaI |
Modified TrEGI + TrEGII + Nourseothricin2 |
|
Strain 2
|
1. BbsI and SmaI 2. NcoI and SalI 3. SalI and NcoI 4. SmaI |
Modified PfCBHI + NcCBHI + TrCBHII + Nourseothricin |
|
Strain 3
|
1. SmaI and BbsI 2. SmaI |
NpBGS + Nourseothricin |
1Digest with these enzymes to expose the appropriate Gibson homology sequence to create the desired plasmid in column three.
2Nourseothricin= Nourseothricin resistance gene
The resulting Gibson Assembled plasmids will be transformed into E. coli for propagation and plasmid amplification. Following extraction, the plasmids will then be linearized with NotI, gel purified, and each will be transformed into a Y. lipolytica competent cell aliquot using the lithium acetate method to create the three strains.
If these gene cassettes are unable to be made after several attempts and troubleshooting to change the DNA concentrations/reaction conditions, then these cassettes can be assembled in many different ways, including sequentially. Check out the appendix page and the part collection page to learn more or see the different kinds of plasmids that can be produced with our collection.
We were very fortunate to be given protocols for transformation of DNA into Y. lipolytica by Dr. Rodrigo Ledesma-Amaro. As per this protocol, cells will be made chemically competent using lithium acetate and transformed with the linearized gene cassettes containing nourseothricin as a selection marker. If the transformation is successful, we expect colonies to develop when plated on YPD with nourseothricin. If this method does not work upon changing the concentration of DNA or incubation periods, other protocols using electroporation may give higher success rates.
Colonies that appear on the nourseothricin plate will undergo colony PCR (cPCR) to verify the integration of the insert into the genome. The parts were designed to be verified using the following primers:
Forward Primer: 5’ GCTTGCTAATGTTAAGTCTCTGC 3’
Reverse Primer: 5’ GGATTCACATCAGTCAATCACC 3’
We will run the PCR products on a 1% gel electrophoresis. The virtual gel below shows the expected results of the gel.

Figure 6. Virtual gel showing the results of a Yarrowia lipolytica cPCR upon transformation with the cellulase gene cassettes. Strain 1 contains Modified TrEGI + TrEGII + Nourseothricin resistance (expected band size= 6187bp), strain 2 contains Modified PfCBHI +NcCBHI + TrCBHII + Nourseothricin resistance (expected band size= 9595bp), and strain 3 contains NpBGS + Nourseothricin resistance (expected band size= 4529bp).
SDS PAGE gels provide a qualitative verification of protein production. We will use them to confirm that our cellulase enzymes are being produced and secreted successfully. To do so, we will run both cell lysate and supernatant on the gels to determine whether the enzymes are being secreted into the extracellular solution as intended. A 10% SDS PAGE will be run with the following expected results:
Table 2. Expected band sizes seen on a 10% SDS page for engineered Yarrowia lipolytica strains 1, 2, and 3. Strain 1 contains Modified PfEGI + TrEGII + Nourseothricin resistance, strain 2 contains Modified PfCBHI + NcCBHI + TrCBHII + Nourseothricin resistance, and strain 3 contains NpBGS + Nourseothricin resistance. Two negative controls include Y. lipolytica transformed with only Nourseothricin resistance, and untransformed Y. lipolytica
|
Sample |
Expected Band for Cell Lysate |
Expected Band for Supernatant |
|
Strain 1
|
Bands at:
|
Large bands: at
|
|
Strain 2
|
Bands at:
|
Large bands at:
|
|
Strain 3
|
Bands at:
|
Large bands at:
|
|
Negative control: Y. lipolytica + Nourseothricin |
Band at 20.4 kDa |
No bands corresponding to our recombinant proteins |
|
Negative control: Untransformed Y. lipolytica |
No bands corresponding to our recombinant proteins |
No bands corresponding to our recombinant proteins |
Contrary to an SDS PAGE gel, an ELISA assay will provide a quantitative measure of protein production. One again, samples of supernatant will be compared to cell lysate samples to see how well our secretion tag is functioning.
Since each strain (1-3) is producing multiple cellulase proteins, each supernatant/cell lysate sample will need to be tested with multiple antibodies in an ELISA assay to determine the concentration of each recombinant protein present in the sample. As per our part design, each cellulase gene within a cassette has a different affinity tag that can be used for antibody binding so that a cellulase-specific antibody is not required. Across all three strains, the cell lysate samples are expected to have less protein than the supernatant sample due to the secretion tag.
Table 3. Experimental design of an ELISA assay to determine how much protein from each engineered Yarrowia lipolyticastrain (1-3) is being produced. Once again, this assay will be run on both cell lysate and supernatant samples from each strain.
|
Sample |
Antibody used1 |
|
Strain 1
|
Antibody=
|
|
Strain 2
|
Antibody=
|
|
Strain 3
|
Antibody=
|
|
Negative control: Y. lipolytica + Nourseothricin |
Antibodies= All Anti-6x His, anti-Myc, and Anti-FLAG antibodies *No antibodies are expected to bind, therefore there should be no fluorescence |
|
Negative control: Untransformed Y. lipolytica |
Antibodies= All Anti-6x His, anti-Myc, and Anti-FLAG antbodies *No antibodies are expected to bind, therefore there should be no fluorescence |
1The antibody being used binds to the affinity tag present on each cellulase protein.
By quantifying the amount of protein being produced, controlled experiments characterizing the cellulases and their activity can be conducted, as they require known amounts of protein.
CELLULASE CHARACTERIZATION
In order to ensure that our cellulases are functioning properly, we intend to conduct individual characterization experiments for each cellulase enzyme. To do so, each protein will be purified, quantified, (by the Bradford Assay) and tested on a different purified substrate to determine the quantity of substrate that is broken down per mg of cellulase enzyme.
Endoglucanases will be tested with carboxymethyl cellulose (CMC) as its substrate (Cano-Ramirez et al, 2016). Beta-glucosidase will be tested with p-nitrophenyl-β-D-glucopyranoside (PNPG) as its substrate (Karnchanatat et al, 2007). Cellobiohydrolases will be tested with Avicel as its substrate (Li et al, 2006). A DNS assay would be able to quantify the amount of activity by way of measuring reducing sugar concentration. Furthermore, BGS activity can be directly tested with a glucose test.
Supernatant samples can also be tested to see how well the cellulases would function in the extracellular media. In this case, ELISA could be used to quantify the protein sample.
TESTING CELLULASES TOGETHER
We can then take samples of the extracellular media, combine them together, and mix with cellulose-based media to characterize the amount of cellulose degradation occurring when all enzymes are preset together. Through DNS assay or a glucose test would measure the glucose production. At this point we can see if the amount of each cellulase being produced is enough based on our preliminary cellulose tests below, and then go back to our part design to change the signal peptide, promoter, or gene integration location to increase expression, or change the growing conditions to optimize Yarrowia lipolytica growth.
CO-CULTURING
Our next step is to co-culture the strains in the same media and measure the amount of glucose that is produced through a DNS assay or glucose test. The co-culturing experiments will be completed on both pure cellulose and crude cellulose. We will also measure the presence of each strain to ensure that no strain is outcompeting the others. To do this, we can integrate a different reporter genes into each strain and measure the fluorescence at different wavelengths, providing a quantitative measure of the quantity of each strain in the co-culture. For more information on this assay check out our Bioncontainment project
PRELIMINARY CELLULASE TESTING
With all of our experimental plans laid out for genetically engineering Y. lipolytica we wanted to do some preliminary cellulase testing to ensure our overall idea of culturing Y. lipolytica on cellulose material in a low-tech growing vessel would actually work. To do so, we conducted the following experiments to understand the growth requirements of Y. lipolytica, and its ability to grow in a cellulose/cellulase solution.
- Glucose growth testing
- Cellulase activity testing: On pure cullulose and crude plant biomass
- Y. lipolytica growth with cellulase and pure cellulose
- Y. lipolytica growth with cellulase and crude plant biomass
The minimum amount of glucose required to successfully grow Y. lipolytica is an important threshold to note, as this is the minimum amount the cellulases would have to be producing to maintain a viable culture of Y. lipolytica in the bioreactor. If during co-culturing experiments of the engineered strains, the co-culture is not surviving, potentially the cellulases are not producing enough glucose as determined by this preliminary test. The protocol for this experiment can be found here. Briefly, Y. lipolytica cells were grown in 4 concentrations of glucose/water media, and the OD600 values were recorded over 5 days.

Figure 7. Growth of Yarrowia lipolytica in different concentrations of glucose solution over 120 hours. Trend line equation for 2% glucose is y=0.0044x + 0.0897, trend line equation for 0.2% glucose is y= 0.0054x + 0.0265, trend line equation for 0.02% glucose is y= 0.0085x - 0.1255, and finally the trend line equation for 0.002% glucose is y= 0.0073x +0.1019. Represents the results of two replicates.
The results from this test show that Y. lipolytica can be maintained even at very low glucose concentrations of 0.02 and 0.002% which approximates to 0.02mg/mL. In fact, we were surprised to see the best growth in 0.02% glucose solution, we believe this might be because Y. lipolytica starts accumulating more lipids under scarce nutrient conditions (Sestric et al. 2014) which may have skewed the absorbance values for the lower glucose concentration. This may also explain why the growth decreased at 48 hours as the cells were likely adapting to the limited glucose concentrations.
This is an important find, as even if the cultures have minimal glucose available in the bioreactor, the cells are still able to survive. In fact, they may even be producing more lipids which is beneficial as lipids promote successful vitamin A uptake.
It was important for us to conduct a small-scale lab proof of concept to verify that our cellulase integration idea was sound. Before we grew the yeast, we determined the correlation between cellulase enzyme concentration and glucose production, as this value would inform our genetically engineered yeast in terms of setting the threshold for the concentration of cellulase needed to be produced. We created a standard curve as seen in figure 8 which can be further interpreted by using our glucose standard curve in the appendix.

Figure 8. DNS assay conducted on pure 1% w/v cellulose solutions incubated with different concentrations of pure cellulase. Trend line equation is y=0.3859x + 0.9053.
From our glucose standard, we determined that the A540=1.524[glucose] - 0.2514 (y=1.5424 -0.2514).
To test the growth of Y. lipolytica on pure and crude cellulose this protocol was followed. Briefly, in culture tubes Y. lipolytica cells were mixed with 1% w/v pure cellulose solution or 0.2g of mashed/boiled crude cellulose. Different concentrations of the pure commercial cellulase were added to the solutions. The tubes were incubated at 30ºC shaking over 5 days, and the OD600 measurement was taken every 2 days. Figure 9 shows the growth of Y. lipolytica on pure commercial cellulose, lemongrass, and chickpea shells.
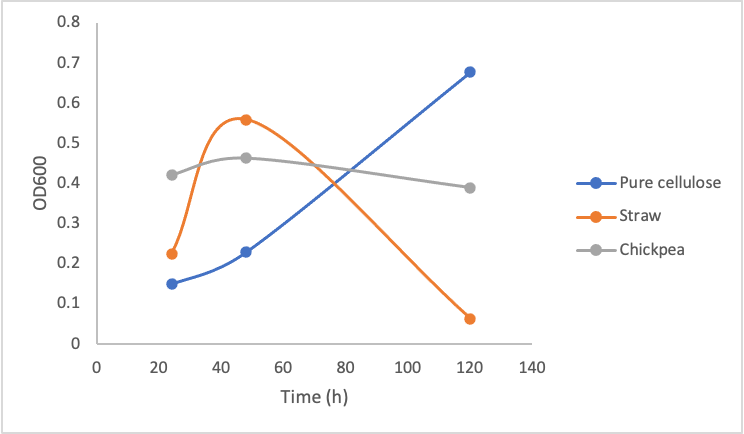

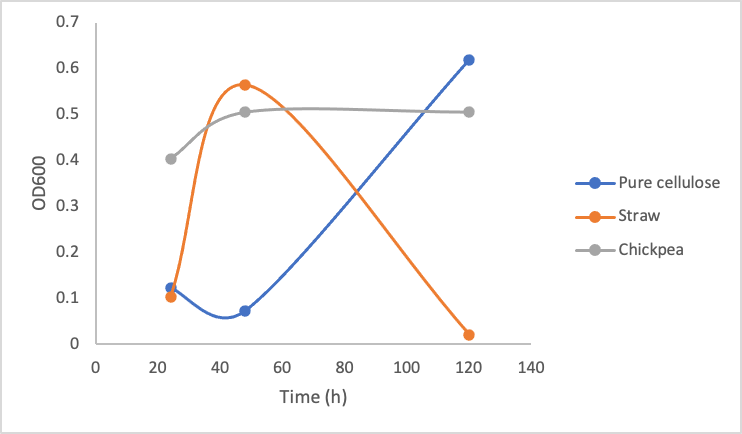
Figure 9. Optical Density (600 nm) of Yarrowia lipolytica over time cultured on pure cellulose, straw, and chickpea shells in the presence of 10 mg/mL (left), 5mg/mL (middle), and 1mg/mL (right) commercial cellulase mixture.
These three graphs do not show significant differences between each other indicating that enzyme concentration did not make a big difference in the growth of the Y. lipolytica cultures. Upon calculating the amount of glucose produced by even 1mg/mL cellulase solution based on figure 8, 1mg/mL of cellulase is able to produce ~1mg/mL of glucose. According to our glucose growth testing results (figure 6), this is 50x greater than the minimal glucose concentration Y. lipolytica cultures were able to grow on. Taking all of these results together, it makes sense that increasing enzyme concentration did not make a difference in Y. lipolytica growth on cellulose material, as the lowest enzyme concentration (1mg/mL) was enough to produce sufficient amounts of glucose.
The most important take away from this experiment is the dramatic drop in growth of the Y. lipolytica cultures with lemongrass and chickpeas shells after a couple days of growth. We do not suspect our enzymes became non-functional, as the cultures in pure cellulose solution continued growing. We suspect the drop in growth occurred due to a decrease in accessibility for the cellulases to the cellulose. When the cultures were first started, the cellulose was boiled and mashed, this likely produced a small amount of slightly degraded and more accessible cellulose for the cellulase to degrade. As time went on, it is possible the shaking incubator did not move the cellulose around enough for sufficient enzymatic activity.
Furthermore, the plant material we used was primarily dry matter, which would consist more of lignin and not cellulose; therefore using more fleshy plant material and having a better pre-treatment process (such as using a grinder/blender) could sustain a Y. lipolytica culture better. Changing the plant material every few days may also improve cellulase activity.
As we move towards genetically engineering our strains and co-culturing our strains together, we should keep these results in mind in terms of the minimal amount of cellulase and glucose production required to sustain cultures, and how the cellulose might need to be continually attended to every few days to make sure it is being efficiently degraded by the cellulases in solution.
FUTURE DIRECTIONS
What we've achieved this year
✔ Selected, modeledl, and optimized our cellulase proteins for function in Y. lipolytica
✔Contributed 20 parts to the iGEM registry
✔Extensively planned all of our experiments for the next year
✔Determined minimum amount of glucose concentration required to maintain Y. lipolytica
✔Determined approximately how much cellulase enzyme is required to reach that minimum glucose concentration
✔Verified that Y. lipolytica can grow on crude (and pure) cellulose when cellulases are present
We are currently in the process of transforming E. coli DH5α with all of our individual parts so the DNA can be amplified, extracted, and used in Gibson Assembly.
iGEM 2021
Below is our timeline for next year. We recognize this may be a little ambitious; however, we believe even getting to protein confirmation and characterization will be a huge step forward for our Cellulase Integration project.

Iterate and Optimize: Beyond iGEM
By enabling our cells to produce cellulases, our project gains the potential to be a real and cost-effective solution as a locally-grown nutritional yeast. Therefore, Cellulase Integration is a cornerstone of our project design. Beyond even next iGEM season, we intend to optimize our design and further characterize cellulase production and cellulose degradation ability for functionality in our bioreactor system.
Additionally, we plan to characterize the fitness of our yeast after modification compared to the wild type. These experiments will test the heat tolerance, pH tolerance, growth rate, oxygen requirements, and sugar and nutrient consumption of modified Y. lipolytica .
Another major future direction of cellulase engineering will be to integrate production of all cellulase types into one single strain. This will allow us to optimize the balance of secreted cellulase types, and prevent one strain from outcompeting or limiting another in the co-culture.
REFERENCES
Brigham, Christopher. 2018.Chapter 3.22 - Biopolymers: Biodegradable Alternatives to Traditional Plastics, In: Green Chemistry,Editor(s): Béla Török, Timothy Dransfield, Elsevier, Pages 753-770, ISBN 9780128092705, https://doi.org/10.1016/B978-0-12-809270-5.00027-3.
Celińska, E., Borkowska, M., Białas, W., Korpys, P., & Nicaud, J. M. (2018). Robust signal peptides for protein secretion in Yarrowia lipolytica: identification and characterization of novel secretory tags. Applied microbiology and biotechnology, 102(12), 5221–5233. https://doi.org/10.1007/s00253-018-8966-9
Guo, Z., Duquesne, S., Bozonnet, S. et al. (2017) Conferring cellulose-degrading ability to Yarrowia lipolytica to facilitate a consolidated bioprocessing approach. Biotechnol Biofuels, 10, 132. https://doi.org/10.1186/s13068-017-0819-8
Niehus X, Crutz-Le Coq AM, Sandoval G, Nicaud JM, Ledesma-Amaro R. Engineering Yarrowia lipolytica to enhance lipid production from lignocellulosic materials. Biotechnol Biofuels. 2018 Jan 22;11:11. doi: 10.1186/s13068-018-1010-6. PMID: 29387172; PMCID: PMC5776775.
Sestric, R., Munch, G., Cicek, N., Sparling, R., & Levin, D. B. (2014). Growth and neutral lipid synthesis by Yarrowia lipolytica on various carbon substrates under nutrient-sufficient and nutrient-limited conditions. Bioresource technology, 164, 41–46. https://doi.org/10.1016/j.biortech.2014.04.016
Taylor, L.E., Knott, B.C., Baker, J.O. et al. (2018). Engineering enhanced cellobiohydrolase activity. Nat Commun, 9, 1186 https://doi.org/10.1038/s41467-018-03501-8
Wang, X., Rong, L., Wang, M., Pan, Y., Zhao, Y., & Tao, F. (2017). Improving the activity of endoglucanase I (EGI) from Saccharomyces cerevisiae by DNA shuffling. RSC Adv., 7, 46246-46256. doi:https://doi.org/10.1039/C6RA26508A
Wei, H., Wang, W., Alper, H. S., Xu, Q., Knoshaug, E. P., Van Wychen, S., Lin, C. Y., Luo, Y., Decker, S. R., Himmel, M. E., & Zhang, M. (2019). Ameliorating the Metabolic Burden of the Co-expression of Secreted Fungal Cellulases in a High Lipid-Accumulating Yarrowia lipolytica Strain by Medium C/N Ratio and a Chemical Chaperone. Frontiers in microbiology, 9, 3276. https://doi.org/10.3389/fmicb.2018.03276



