Team:GunnVistaPingry US/Model
Training and test datasets
The training data set was randomly divided into 17 gene folds where each fold has a further distribution of 94% and 6% for training and test data sets, respectively.
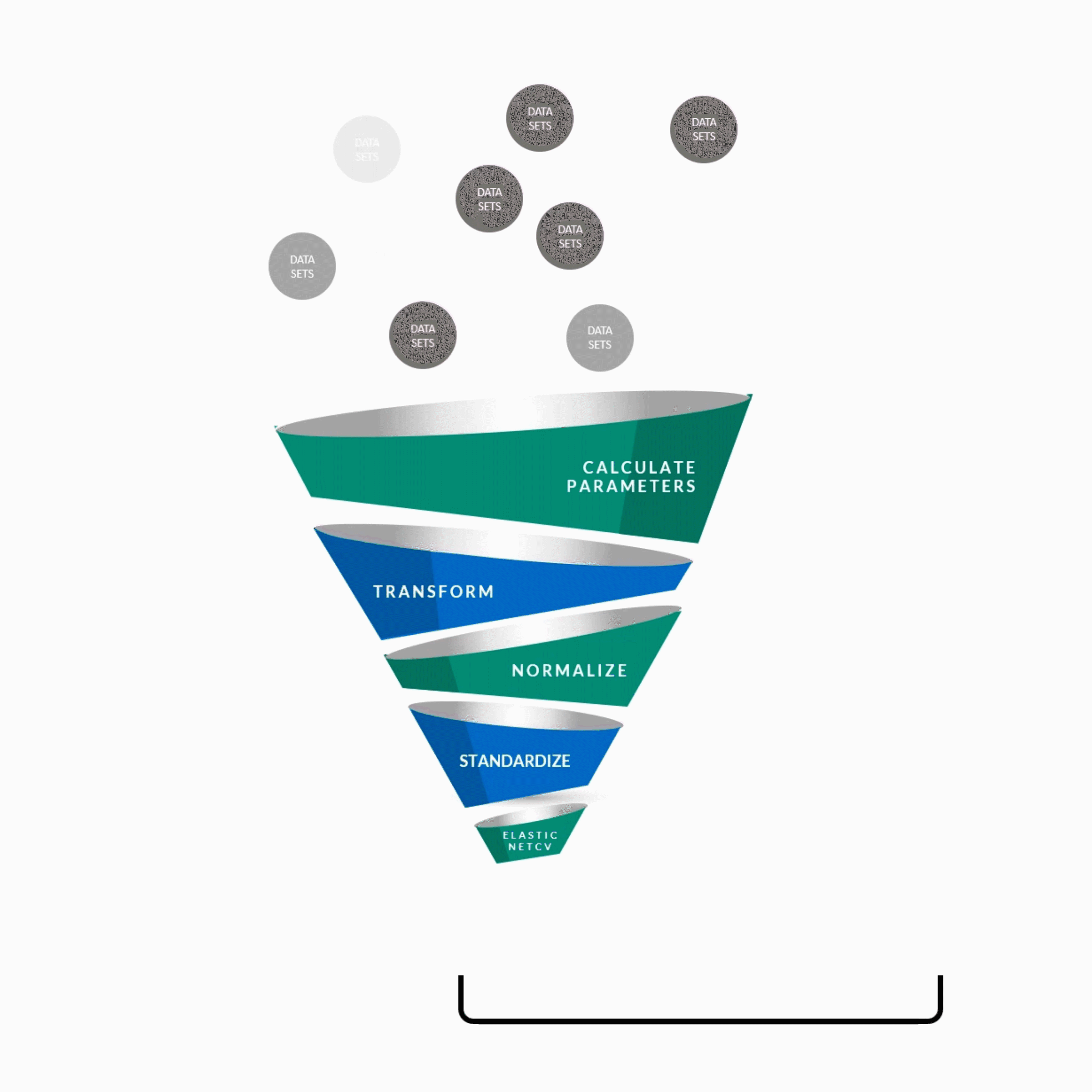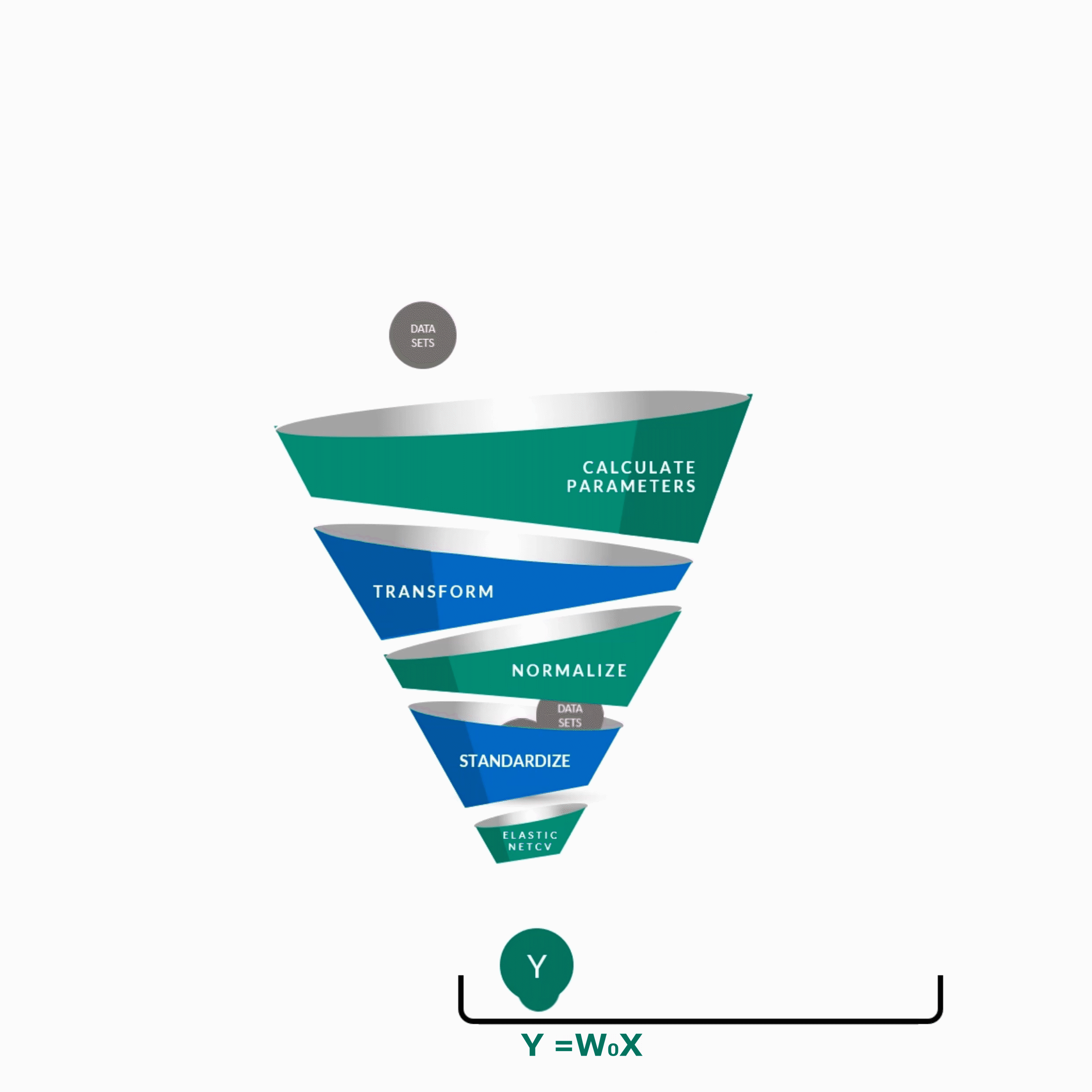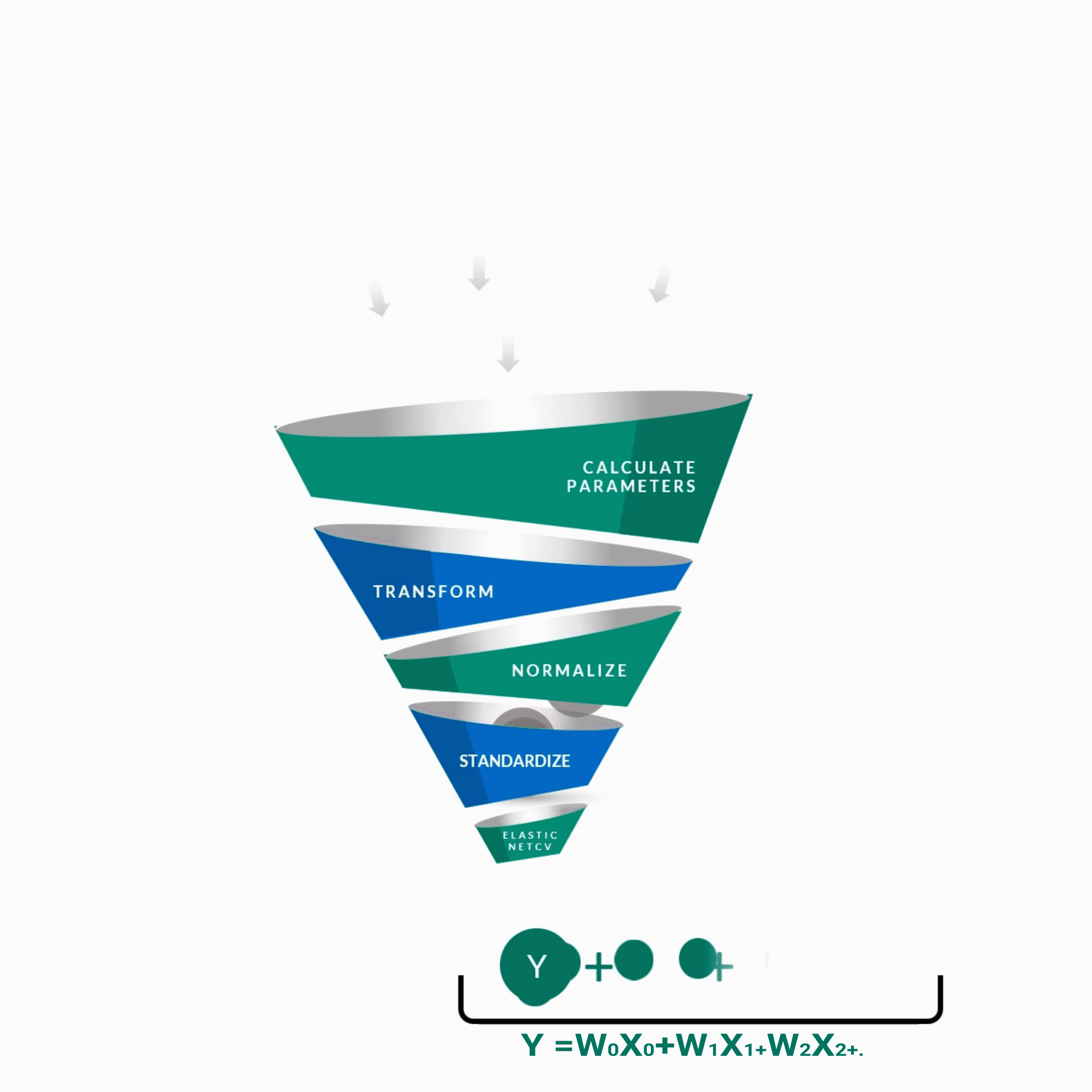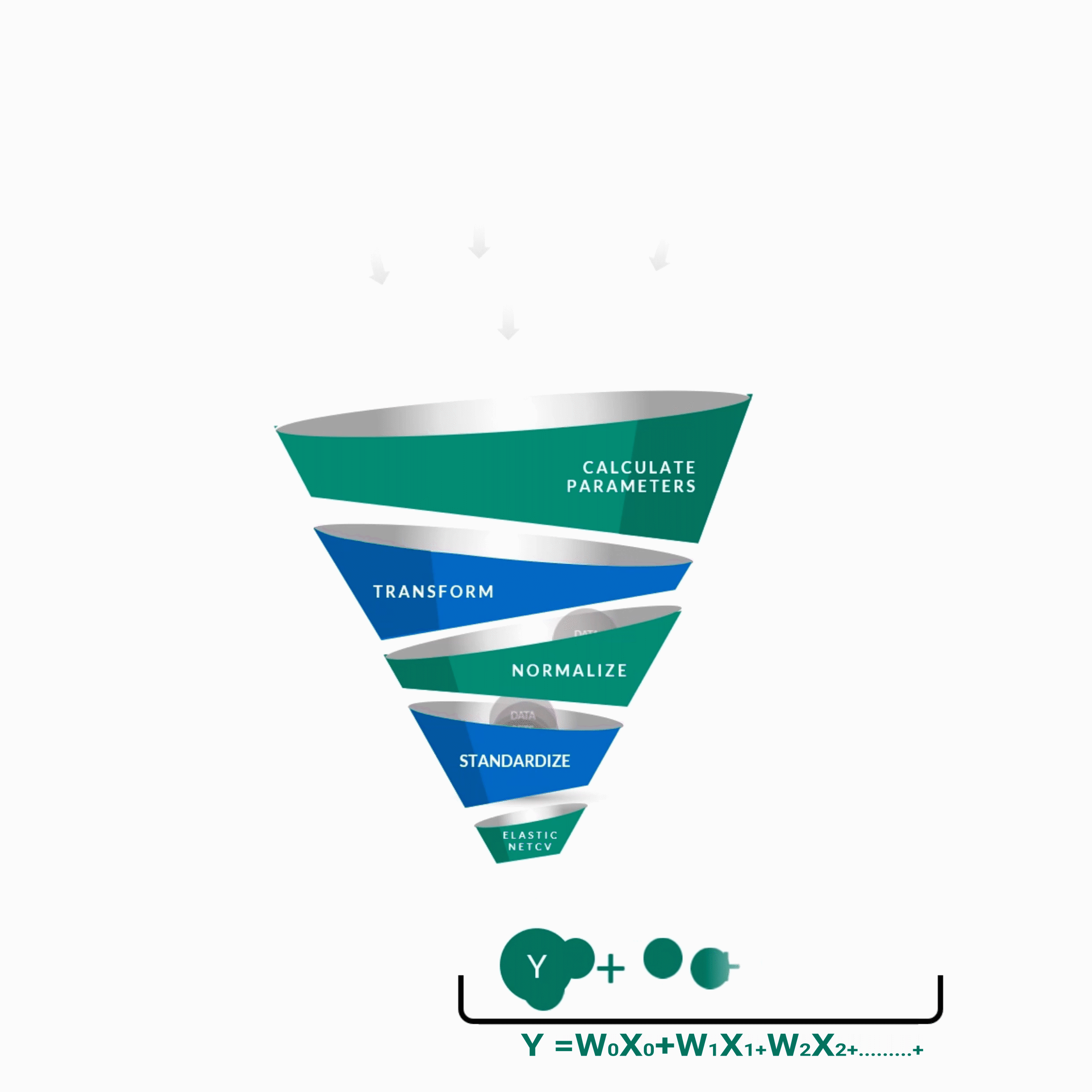
y = w0x0 + w1x1 + w2x2 + w3x3 + w4x4 + ................. +wnxn
y -- Score of the Guide
w0 ... wn -- Weights of features contributing to score
x0 ... xn -- Features contributing to score as predicted by the algorithm.
Features
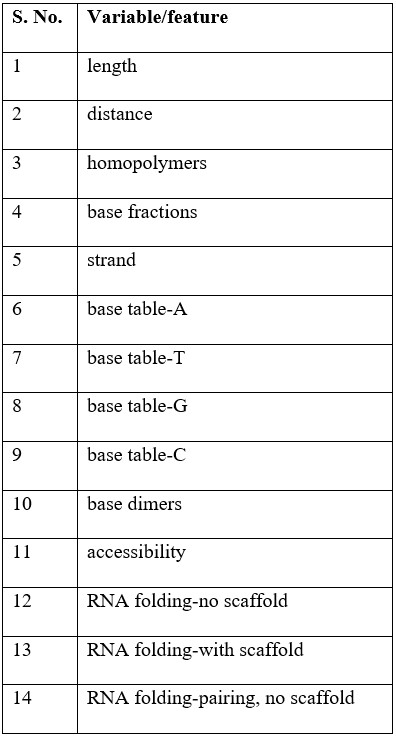
Table-1: List of variables/features
Transformation process[1]
Parameters were binned by a fixed width for each feature and applied over the range of values, with the upper-most and lower-most bins collapsed with the neighboring bins if the number of data points at each edge were sparse.
For binning parameters, a fixed-width was chosen for each feature/parameter and applied over the range of values, with the upper-most and lower-most bins collapsed with the neighboring bins if the number of data points at each edge were sparse. Each feature was then split into individual parameters for each bin and sgRNAs were assigned a 1 for the bin if the value fell within the bin or 0 if not. For linearizing sgRNA positioning parameters with continuous curves, sgRNA positions were fit to the activity score (individually for the distance to each TSS coordinate) using SVR with a radial basis function kernel and hyperparameters C and gamma determined using a grid search approach cross-validated within the training set. The fit score at each position was then used as the transformed linear parameter. Binary parameters were assigned a 1 if true or a 0 if false. All linearized parameters were z-standardized and fit with elastic net linear regression, with different L1/L2 ratio values [.5, .75, .9, .99, 1] as suggestions.
Calculating features[2]
ViennaRNA package was used with default parameters to calculate RNA folding metrics. The signal at each base of the target site including the PAM was averaged to calculate the chromatin features. Bx-python module was used to extract the processed continuous signal from the following BigWig files obtained from the ENCODE Consortium: MNase-seq https://www.encodeproject.org/files/ENCFF000VNN/ (Michael Snyder lab, Stanford University), DNase-seq https://www.encodeproject.org/files/ENCFF000SVY/ (Gregory Crawford lab, Duke University), and FAIRE-seq https://www.encodeproject.org/files/ENCFF000TLU/ (Jason Lieb lab, University of North Carolina) (ENCODE Project Consortium, 2012).
Method
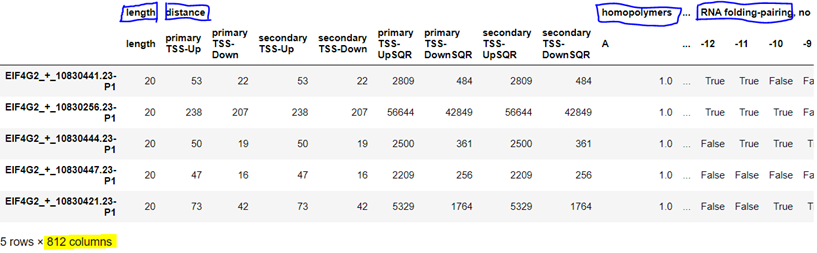
Fig-1: The top-level feature “distance” was translated into 8 low-level features: primary and secondary upstream/downstream normal distances & squares of distances from TSS
Note: ElasticNetCV is a combination of Lasso and Ridge regressions. Lasso takes care of data set’s collinearity issues and ridge makes sure the regression model is not overfitted.
Findings
Note: The coefficient R^2 is defined as (1 - u/v), where u is the residual sum of squares ((y_true - y_pred) ** 2).sum() and v is the total sum of squares ((y_true - y_true.mean()) ** 2).sum(). The best possible score is 1.0 and it can be negative (because the model can be arbitrarily worse). A constant model that always predicts the expected value of y, disregarding the input features, would get a R^2 score of 0.0.
[https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.ElasticNetCV.html]
As per the linear model that was generated, the following features (Table-1) contributed to the target activity score with corresponding coefficients (Fig-2).
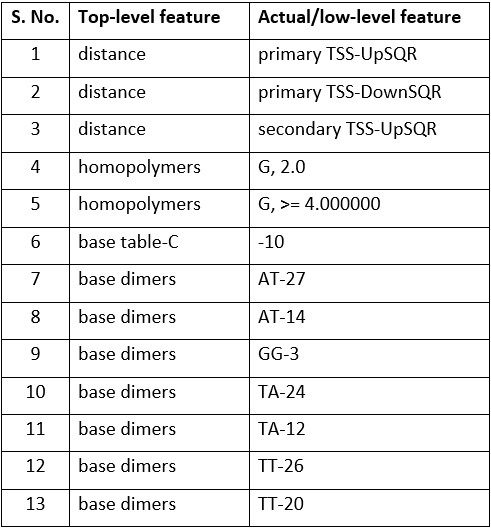
Table-2: Low-level variables/features of regression model with highest R 2 value.
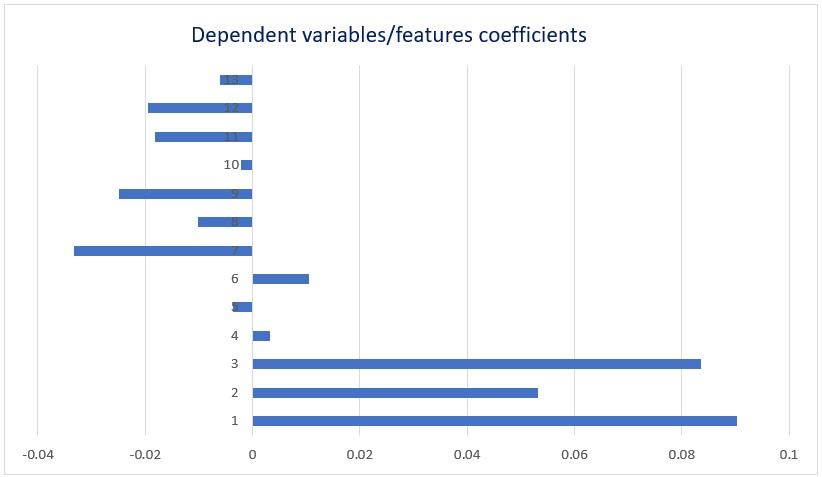
Fig-2: Coefficients of low-level variables/features of regression model with highest R 2 value.
Discussion
Click here for the results.
Scatter plots
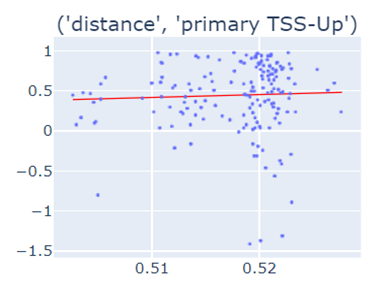
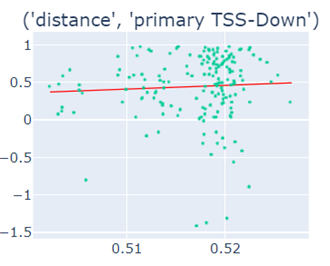
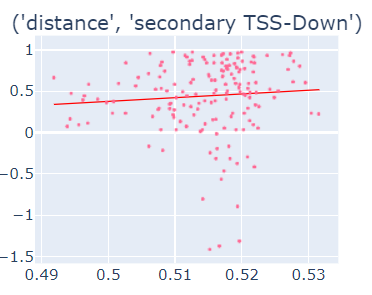
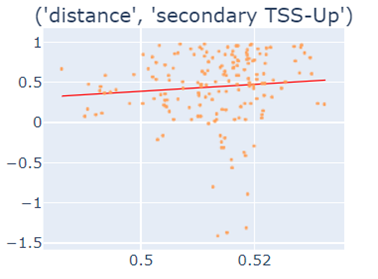
Fig-3: Scatter plots of primary/secondary upstream/downstream distances from TSS vs our algorithm score.
However, when we used squares of primary/secondary upstream/downstream distances from TSS to guide, we identified a strong & clear linear relationship.
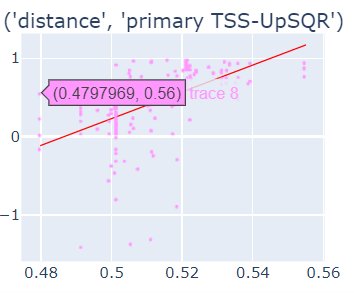

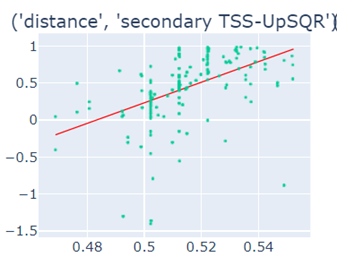
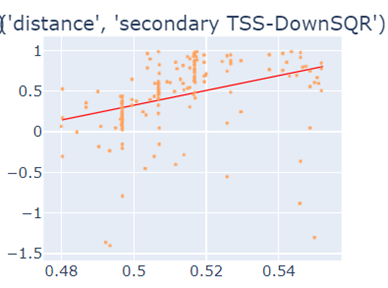
Fig-4: Scatter plots of squares of primary/secondary upstream/downstream distances from TSS vs our algorithm score.
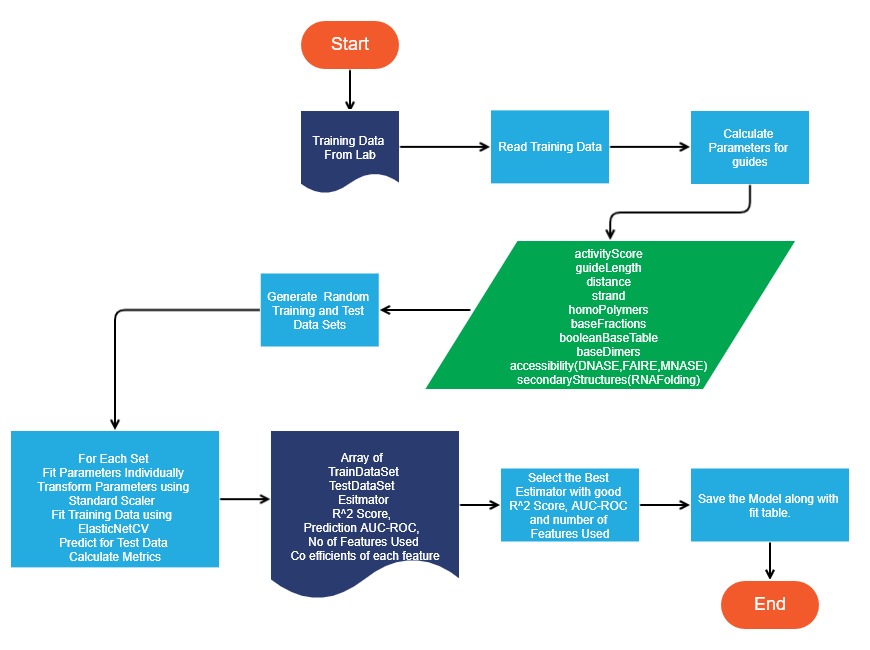
Model Generator Flowchart
Limitations
- Availability of large quantity of qRT-PCR training data is difficult to obtain than phenotype based data
- Model is trained based on human genome hg19
Acknowledgment: Our model was inspired from https://elifesciences.org/articles/19760 research paper. We extend our sincere gratitude to all the authors & contributors of the paper.
